RNA Seq Read Representation by Trinity Assembly
Assembled transcripts might not always fully represent properly paired-end reads, as some transcripts may be fragmented or short and only one fragment read of a pair may align. Simply aligning reads to your transcriptome assembly using bowtie or STAR will only capture the properly paired reads. To assess the read composition of our assembly, we want to capture and count all reads that map to our assembled transcripts, including the properly paired and those that are not.
In order to comprehensively capture read alignments, we run the process below. Bowtie2 is used to align the reads to the transcriptome and then we count the number of proper pairs and improper or orphan read alignments.
% bowtie2-build Trinity.fasta Trinity.fasta
Example for paired-end reads:
% bowtie2 -p 10 -q --no-unal -k 20 -x Trinity.fasta -1 reads_1.fq -2 reads_2.fq \
2>align_stats.txt| samtools view -@10 -Sb -o bowtie2.bam
Example for single-end reads:
% bowtie2 -p 10 -q --no-unal -k 20 -x Trinity.fasta -U single.reads.fq \
2>align_stats.txt| samtools view -@10 -Sb -o bowtie2.bam
% cat 2>&1 align_stats.txt
The output bellow:
76201190 reads; of these:
76201190 (100.00%) were paired; of these:
18166307 (23.84%) aligned concordantly 0 times
17026716 (22.34%) aligned concordantly exactly 1 time
41008167 (53.82%) aligned concordantly >1 times
----
18166307 pairs aligned concordantly 0 times; of these:
1769907 (9.74%) aligned discordantly 1 time
----
16396400 pairs aligned 0 times concordantly or discordantly; of these:
32792800 mates make up the pairs; of these:
15287552 (46.62%) aligned 0 times
3874965 (11.82%) aligned exactly 1 time
13630283 (41.56%) aligned >1 times
89.97% overall alignment rate
A typical Trinity transcriptome assembly will have the vast majority of all reads mapping back to the assembly, and ~70-80% of the mapped fragments found mapped as proper pairs (yielding concordant alignments 1 or more times to the reconstructed transcriptome).
Note, if you see high rates of duplication, usually this is not a cause for concern. Highly expressed transcripts with alternatively spliced isoforms will account for multiple read mappings. Later, abundance estimation (transcript expression quantification) takes these multiple mappings into account.
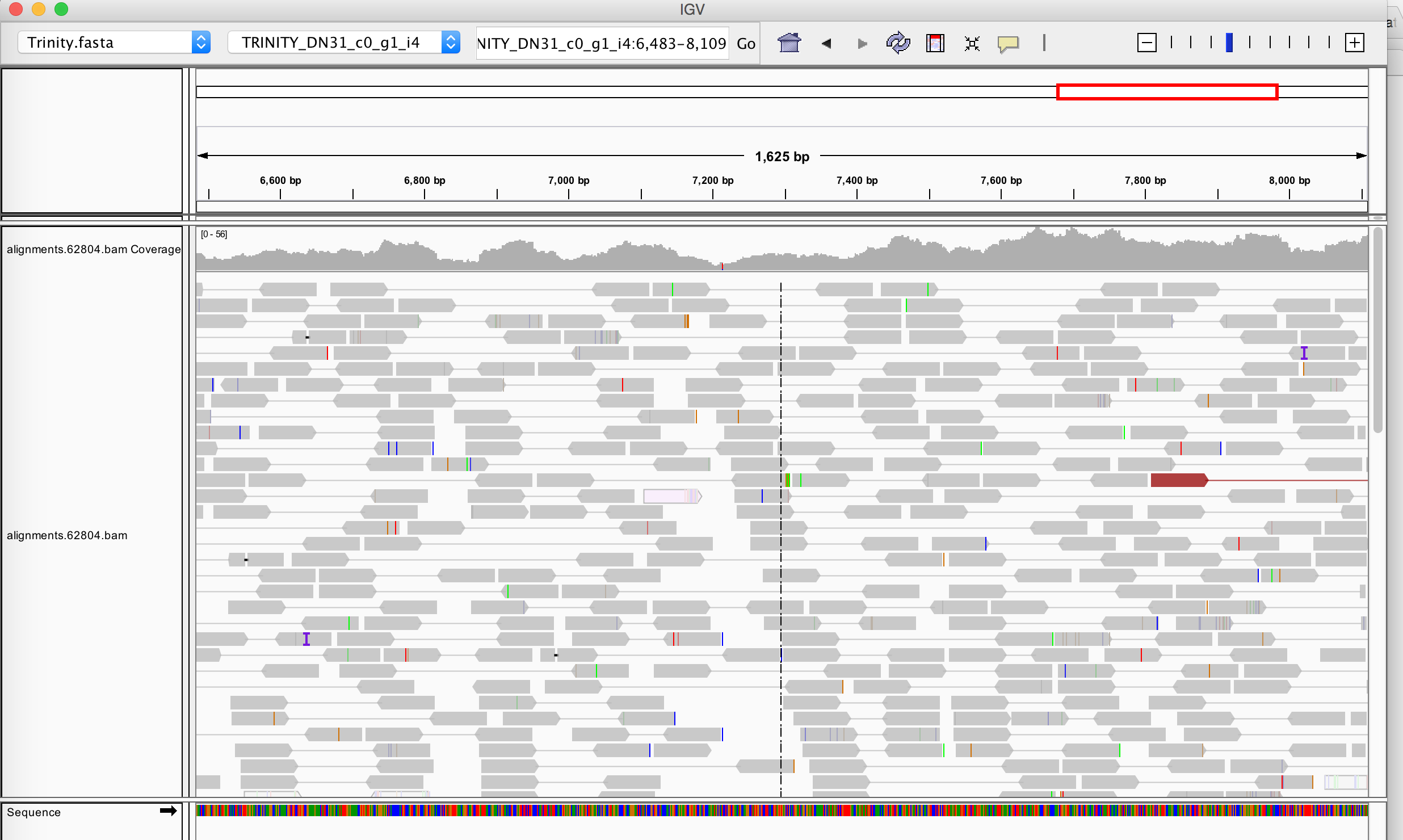
The Integrative Genomics Viewer is useful for visualizing read support across any of the Trinity assemblies. The bowtie2 alignments generated above, which are currently sorted by read name, can be re-sorted according to coordinate, indexed, and then viewed along with the Trinity assemblies using the IGV browser as follows.
samtools sort bowtie2.bam -o bowtie2.coordSorted.bam
samtools index bowtie2.coordSorted.bam
samtools faidx Trinity.fasta
note, you can do this by using the various graphical menu options in IGV (load genome 'Trinity.fasta', load file 'bowtie2.coordSorted.bam'), or you can use the command-line tool like so:
igv.sh -g Trinity.fasta bowtie2.coordSorted.bam
Note, the above assumes that the Trinity.fasta and bowtie2.coordSorted.bam files are in your current working directory.
And you can then go to any Trinity assembly of interest and examine the read (and paired-end) support. An example region of a long Trinity transcript contig is shown below.