Releases: davidemms/OrthoFinder
OrthoFinder v2.5.5
New in this release
- Reduce number of open files when writing orthologs to approximately one per species instead of one per species-pair, this should resolve issues related to ulimit.
- Added option
--fewer-files: Requests that OrthoFinder only write one orthologs file per species. This file will list all orthologs in all other species (the default is one file of orthologs for each species pair, listing only the orthologs between those two species).
-- Added scriptscripts_of/split_ortholog_files.pyto recreate one file of orthologs per species-pair from a OrthoFinder results directory produced with the--fewer-filesoption. - Dependency checks: Print debug info & preserve test files if dependency checks fail for tools that OrthoFinder calls.
Fixes:
OrthoFinder v2.5.4
New in this release
- Add tool create_files_for_hogs.py for creating sequence fasta files for HOGs
- Extend primary_transcripts.py script to interpret NCBI files
- Reduce RAM usage when trimming for very large alignments
- Resolve #526: Handle multiprocessing error occurring only in old versions of glibc
- Resolve #557: Progress reports were sometimes reported out of order
- Resolve #567: Check that the requested number of threads is positive
- Resolve #570: Use fork instead of spawn on Mac
- Resolve #580: Fix to allow primary transcripts script to work for NCBI isoforms labelled with letters
- Resolve #586: Use tempfile library to handle tmp folders
- Fix a problem with overwriting MSA files
OrthoFinder v2.5.2
New in this release
Added option to use DIAMOND ultra-sensitive: "-S diamond_ultra_sens". This identifies homologs for approximately 2% more genes, depending on how closely the input species are related.
OrthoFinder v2.5.1
New in this release
- Significant speed improvements for large analyses
- For analyses of ~200 species total run times are 2-4x faster
- Parallelisation of final ortholog inference stage of algorithm (number of threads is controlled using "-a" option)
- For MSA tree inference OrthoFinder performs light trimming of the MSA. This prevents the runtime being dominated by tree inference for the largest orthogroups with very gappy MSAs.
- The tree inference using multiple sequence alignments option ("-M msa") is now comparable in speed to the default DendroBLAST method.
OrthoFinder v2.4.1
New in this release
- Improvements to the accuracy of phylogenetically inferred hierarchical orthogroups (HOGs)
- Allow
config_orthofinder_user.jsonas an extra config file in user's home directory to allow user-specific options and carrying user options between releases - Allow analysis of nucleotide sequences with
-doption - Resolve #453
- Resolve #475
- Resolve #476
Details
- Orthogroups are now inferred using gene trees and are found in Phylogenetic_Hierarchical_Orthogroups/N0.tsv etc. The original OGs inferred using clustering are still in Orthogroups/Orthogroups.tsv, but the N0.tsv orthgroups are ~12% more accurate and should be used instead.
- The accuracy can be increased still further (20% more accurate on Orthobench) by including outgroup species, which help with the interpretation of the rooted gene trees. The species tree should then be used to identify the correct HOG file, N??.tsv according to the correct node of the species tree.
- It is important to ensure that the species tree OrthoFinder is using is accurate so as to maximise the accuracy of the HOGs. To reanalyse with a different species tree use the options
-ft PREVIOUS_RESULTS_DIR -s SPECIES_TREE_FILE. This runs just the final analysis steps "from trees" and is relatively quick. - Further accuracy increases can be obtained by using a lower MCL inflation value (e.g.
-I 1.3) since this brings more genes into the gene trees, and the HOG algorithm will split the hierarchical orthogroups if required. On Orthobench this gives ~2% increase in accuracy.
OrthoFinder v2.4.0
New in this release
Phylogenetically inferred orthogroups: OrthoFinder now creates a new directory that contains orthogroups defined at each level in the species tree. These orthogroups are inferred by examining the gene trees using the same algorithm that OrthoFinder uses to infer orthologs. Because they are inferred by analysing gene trees they are substantially more accurate than any other method available (and give an approximately 10% relative increase in accuracy on the Orthobench benchmarks compared to OrthoFinder version 2). These files are in the new results directory Phylogenetic_Hierarchical_Orthogroups/.
Because OrthoFinder now infers orthogroups at each phylogenetic level within the species tree it is now possible to include outgroup species in your analysis. Then, to see the orthogroups for just your species of interest just use the corresponding file from the Phylogenetic_Hierarchical_Orthogroups/. The clade names N1, N2, etc. can be found in Species_Tree/SpeciesTree_rooted_node_labels.txt. The use of outgroup species can further increase accuracy (~13% relative increase compared to OrthoFinder v2).
Hierarchical orthogroups are useful because, due to gene duplication events, orthogroups become more fine grained as the species become more closely related:
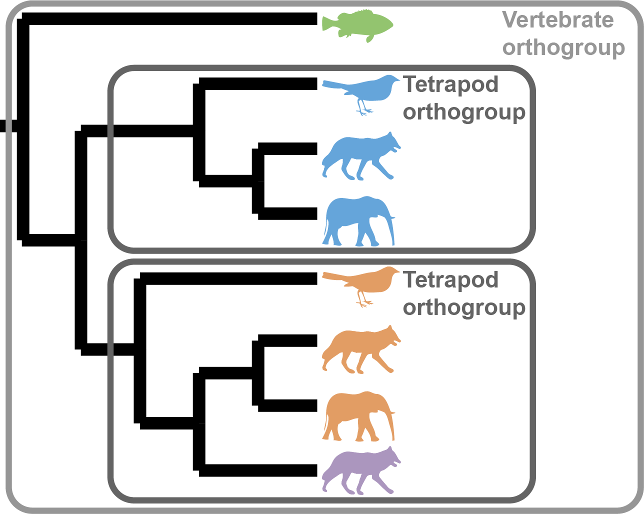
This is the first of a two part series of developments to increase OrthoFinder orthogroup accuracy using the analysis of gene trees.
Which package to download:
- On Linux download OrthoFinder.tar.gz. This bundles all the required external dependencies (mcl, diamond, fastme) and python libraries and so should run immediately, without any installation being required.
- On Mac the bioconda package is probably the easiest method: See Bioconda getting started and, once bioconda is set up, run
conda install orthofinder - On either platform you can run the source code version but you will need to have python and the numpy & scipy libraries installed.
- On Windows the best way is to install the Windows Subsystem for Linux and then use the linux version
More detailed instructions here: https://davidemms.github.io/orthofinder_tutorials/alternative-ways-of-getting-OrthoFinder.html
OrthoFinder v2.3.14 (stable)
This is a stable release that fixes any known issues in the previous release.
Which package to download:
- On Linux download OrthoFinder.tar.gz. This bundles all the required external dependencies (mcl, diamond, fastme) and python libraries and so should run immediately, without any installation being required.
- On Mac the bioconda package is probably the easiest method: See Bioconda getting started and, once bioconda is set up, run
conda install orthofinder - On either platform you can run the source code version but you will need to have python and the numpy & scipy libraries installed.
- On Windows the best way is to install the Windows Subsystem for Linux and then use the linux version
More detailed instructions here: https://davidemms.github.io/orthofinder_tutorials/alternative-ways-of-getting-OrthoFinder.html
Issues resolved
OrthoFinder v2.3.12 (stable)
This is a stable release that fixes any known issues in the previous release.
Which package to download:
- On Linux download OrthoFinder.tar.gz. This bundles all the required external dependencies (mcl, diamond, fastme) and python libraries and so should run immediately, without any installation being required.
- On Mac the bioconda package is probably the easiest method: See Bioconda getting started and, once bioconda is set up, run
conda install orthofinder - On either platform you can run the source code version but you will need to have python and the numpy & scipy libraries installed.
- On Windows the best way is to install the Windows Subsystem for Linux and then use the linux version
More detailed instructions here: https://davidemms.github.io/orthofinder_tutorials/alternative-ways-of-getting-OrthoFinder.html
Issues resolved
- Update primary_transcript.py for python3, resolves #345
- Vectorise alignment trimming, 45mins->1.5s on 6 species x 3 million base alignment
- Updates to Manual & README
- Set OPENBLAS_NUM_THREADS=1, resolves #356
- Fix reporting of external program error messages
- Exception.message deprecated in python3, resolves #375
- Correct handling of species tree without support values, resolves #379
- Improve handling of commented out species
- Check at start if open file limit is too low and inform user, resolves #384
OrthoFinder v2.3.11
Which version to download:
- On Linux download OrthoFinder.tar.gz. This bundles all the required external dependencies (mcl, diamond, fastme) and python libraries and so should run immediately, without any installation being required.
- On Mac the bioconda package is probably the easiest method: See Bioconda getting started and, once bioconda is set up, run
conda install orthofinder - On either platform you can run the source code version but you will need to have python and the numpy & scipy libraries installed.
New in this release
- Resolve an issue in some situations when using OrthoFinder on Mac using bioconda. OrthoFinder would find mcl/diamond but would then be unable to call them when required.
- Binary package (OrthoFinder.tar.gz) is now built for glibc versions 2.15 onwards for wider compatibility
OrthoFinder v2.3.10
- Bug fixes for upgrade to Python 3
- Default package is now built for glibc versions 2.15 onwards for wider compatibility