Scripts to generate random short reads from a given set of contigs, so they can be assembled back (to test assemblers, or genome binning software, or more).
Using the config files and scripts,
- You can generate single reads in a FASTA-formatted output file.
- You can generate paired-end reads in FASTQ-formatted output files. You can define an insert size with a standard deviation.
- You can generate reads from a single FASTA file with a single contig, single FASTA file with multiple contigs, or you can simulate a complex metagenomic mixture using multiple FASTA files each of with with multiple contigs.
- You can specify a desired average coverage for each entry, and define an expected error rate for the sequencer.
The tool (both with respect to its functionality, and design) can be improved dramatically, but we only pushed it as much as what we needed from it. If you need something that is not here, please feel free to open an issue.
Note: You will need anvi'o to be installed on your system. Sorry! Find anvi'o installation instructions here: http://merenlab.org/software/anvio
Say, you have one or more FASTA files each of which contains one or more contigs, and you wish to create a single FASTA file that contains enough number of short reads randomly created from these contigs to meet your expected coverage when they are assembled.
The program gen-single-reads allows you to generate those data.
For a little demonstration there is an examles/files directory in the repository that includes 5 FASTA files:
$ ls examples/files/*fa
examples/files/fasta_01.fa
examples/files/fasta_02.fa
examples/files/fasta_03.fa
examples/files/fasta_04.fa
examples/files/fasta_05.faEach FASTA file is sampled from a different bacterial genome, and contains 5 contigs that are about 7,500nts long.
Here is an example config file you can use with gen-single-reads:
[general]
output_file = short_reads.fa
short_read_length = 100
error_rate = 0.05
[files/fasta_01.fa]
coverage = 100
[files/fasta_02.fa]
coverage = 200
[files/fasta_03.fa]
coverage = 50
[files/fasta_04.fa]
coverage = 150
[files/fasta_05.fa]
coverage = 50For each section except the general section, the config file simply says;
"Generage enough 100nt long short reads from all entries you find in sample/fasta_01.fa, so when they are assembled and short reads mapped back to resulting contigs, the average coverage would be about 100X for each contig in sample/fasta_01.fa. While doing this, introduce some random base errors to meet 0.005 error rate in average. In fact do the same for every other entry in this config file with respect to their required coverage values, and store all resulting reads in short_reads.fa".
Once you have your config file ready, this is how you run it:
$ ./gen-single-reads examples/single-reads-example.ini
fasta_01 w/ 5 contigs : 3,800 reads with 1,870 errors (avg 0.0049) for 10X avg cov.
fasta_02 w/ 5 contigs : 76,000 reads with 37,831 errors (avg 0.0050) for 200X avg cov.
fasta_03 w/ 5 contigs : 19,000 reads with 9,536 errors (avg 0.0050) for 50X avg cov.
fasta_04 w/ 5 contigs : 57,000 reads with 28,489 errors (avg 0.0050) for 150X avg cov.
fasta_05 w/ 5 contigs : 19,000 reads with 9,485 errors (avg 0.0050) for 50X avg cov.
Fasta output ...: short_reads.faThere it is. So in theory, if you assemble short_reads.fa, you should recover contigs that are about 50, 100, 150 and 200X coverage. Just for fun, I used velvet to assemble what is in the short_reads.fa, then used [bowtie2](http://bowtie- bio.sourceforge.net/bowtie2/index.shtml) to map reads back to resulting contigs, and analyzed the mapping.
Here what I did step by step (excuse me for the crudeness of my demo and directly copy-pasting from my command line):
$ mkdir velvet_output/
$ velveth velvet_output/ 17 short_reads.fa
$ velvetg velvet_output/
$ bowtie2-build velvet_output/contigs.fa contigs_ref
$ bowtie2 -f short_reads.fa -x contigs_ref -S output.sam
209000 reads; of these:
209000 (100.00%) were unpaired; of these:
4070 (1.95%) aligned 0 times
204930 (98.05%) aligned exactly 1 time
0 (0.00%) aligned >1 times
98.05% overall alignment rate
$ samtools view -bS output.sam > output.bam
I further analyzed the BAM file in an ad hoc manner with in-house scripts.
Here is the average coverage of each contig in output.bam file:
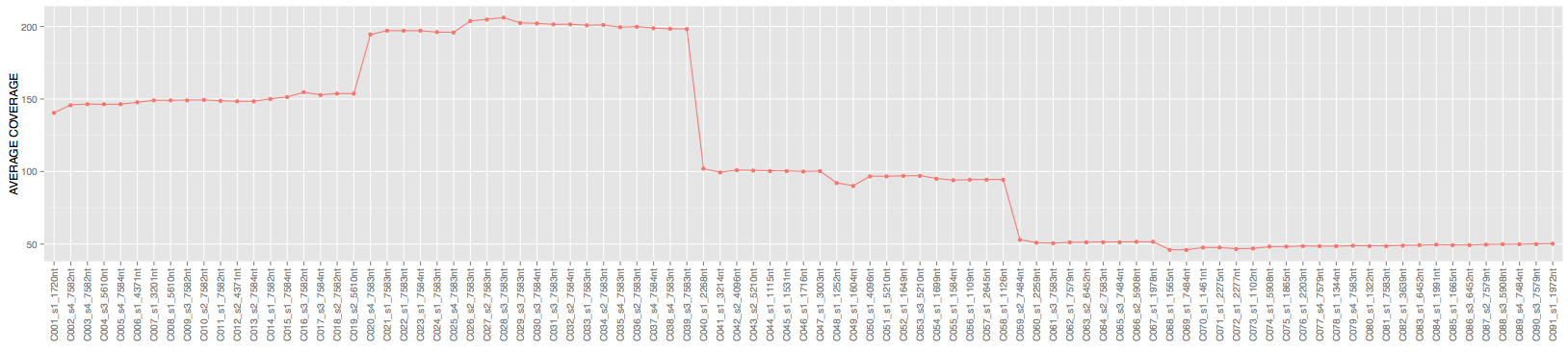
So randomly generated and assembled short reads did creaete expected coverage profiles.
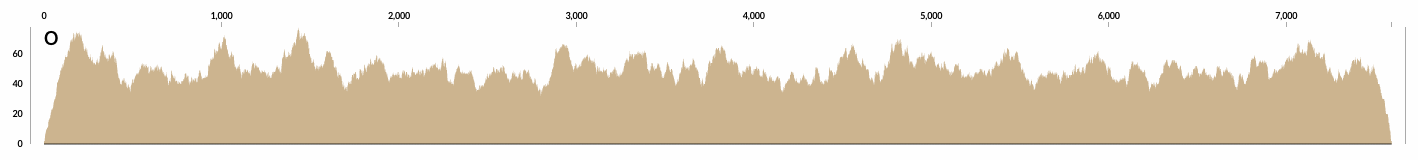
Here is the detailed coverage of one of those contigs that is at 50X coverage:
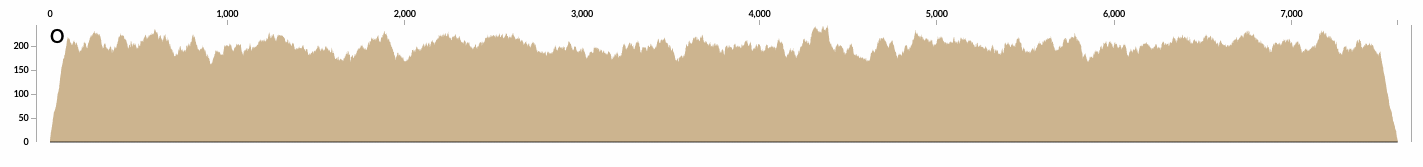
And another example from a contig that is covered about 200X:
Everything is the same, except this time you will use the program gen-paired-end-reads with a config file that looks like this one:
[general]
output_sample_name = test_sample
insert_size = 30
insert_size_std = 1
short_read_length = 100
error_rate = 0.05
[files/fasta_01.fa]
coverage = 10
[files/fasta_02.fa]
coverage = 20
[files/fasta_03.fa]
coverage = 5
[files/fasta_04.fa]
coverage = 15
[files/fasta_05.fa]
coverage = 5insert_size is different than the real insert size, and describes the gap between the end of read 1 and the beginning of read 2.
Here is an example run:
$ ./gen-paired-end-reads examples/paired-end-reads-example.ini
Read lenth ...............: 100
Insert size ..............: 30
Insert size std ..........: 1.0
fasta_01 w/ 5 contigs ....: 3,800 reads in 1900.0 pairs with 1,986 errors (avg 0.0052) for 10X avg cov.
fasta_02 w/ 5 contigs ....: 7,600 reads in 3800.0 pairs with 3,816 errors (avg 0.0050) for 20X avg cov.
fasta_03 w/ 5 contigs ....: 1,900 reads in 950.0 pairs with 924 errors (avg 0.0049) for 5X avg cov.
fasta_04 w/ 5 contigs ....: 5,700 reads in 2850.0 pairs with 2,844 errors (avg 0.0050) for 15X avg cov.
fasta_05 w/ 5 contigs ....: 1,900 reads in 950.0 pairs with 979 errors (avg 0.0052) for 5X avg cov.
FASTQ R1 .................: test_sample-R1.fastq
FASTQ R2 .................: test_sample-R2.fastqAnd this is how these files look like (those A's are made up quality scores :/ it can easily be improved if you need more complex Q-score simulations):
$ head -n 8 test_sample-R*
==> test_sample-R1.fastq <==
@fasta_01:23:B02CBACXX:8:2315:9436:8855 1:N:0:GATCAG
AAAGTGAGCGATACAAAGCTAAAGCCATCAACCAAAAAGCCCGCAACCCGCAAACCAGCTCCGCANACATCGGAGCTATCCAAGGACGATGACTNCCTGT
+source:640612206 slice:0-7600; start:5360; stop:5460; insert_size:32
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
@fasta_01:23:B02CBACXX:8:2315:7851:9246 1:N:0:GATCAG
TGATGAGGCCTTCGTGGCCCAAGAGCGGACCAGCCGTAACTACGACCTGGTCGCCGTCTTTTAGGGCCTCGCTCATGGGCACCACGCGGTTTCCCCTATT
+source:640612206 slice:0-7600; start:2274; stop:2374; insert_size:31
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
==> test_sample-R2.fastq <==
@fasta_01:23:B02CBACXX:8:2315:9436:8855 2:N:0:GATCAG
TAAATTCACCAATGACATGGACACTCTTTACGTCGGGCGCCCAAACGCAAATGCGCCAGCCGCGCGTGCGCTGCTGCGTGTCGGGATTCGCGCCGATCTT
+source:640612206 slice:0-7600; start:5492; stop:5592; insert_size:32
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
@fasta_01:23:B02CBACXX:8:2315:7851:9246 2:N:0:GATCAG
GCGATACGGCAGATCCTCGGACCGTGCAACAGTACCTGCTGCGCATGGATGACTTTGCCCGGGTGCTTTCACAGGACGGTCAGTTTGTGCCGTTGGCAAA
+source:640612206 slice:0-7600; start:2405; stop:2505; insert_size:31
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIt is indeed going to be useful for my purposes, but I would like it to be useful to you as well. So if this is what you need to do benchmarks or to test your stuff, and if its missing something for your purposes, please don't hesitate to get in touch with me.