{kind=link}
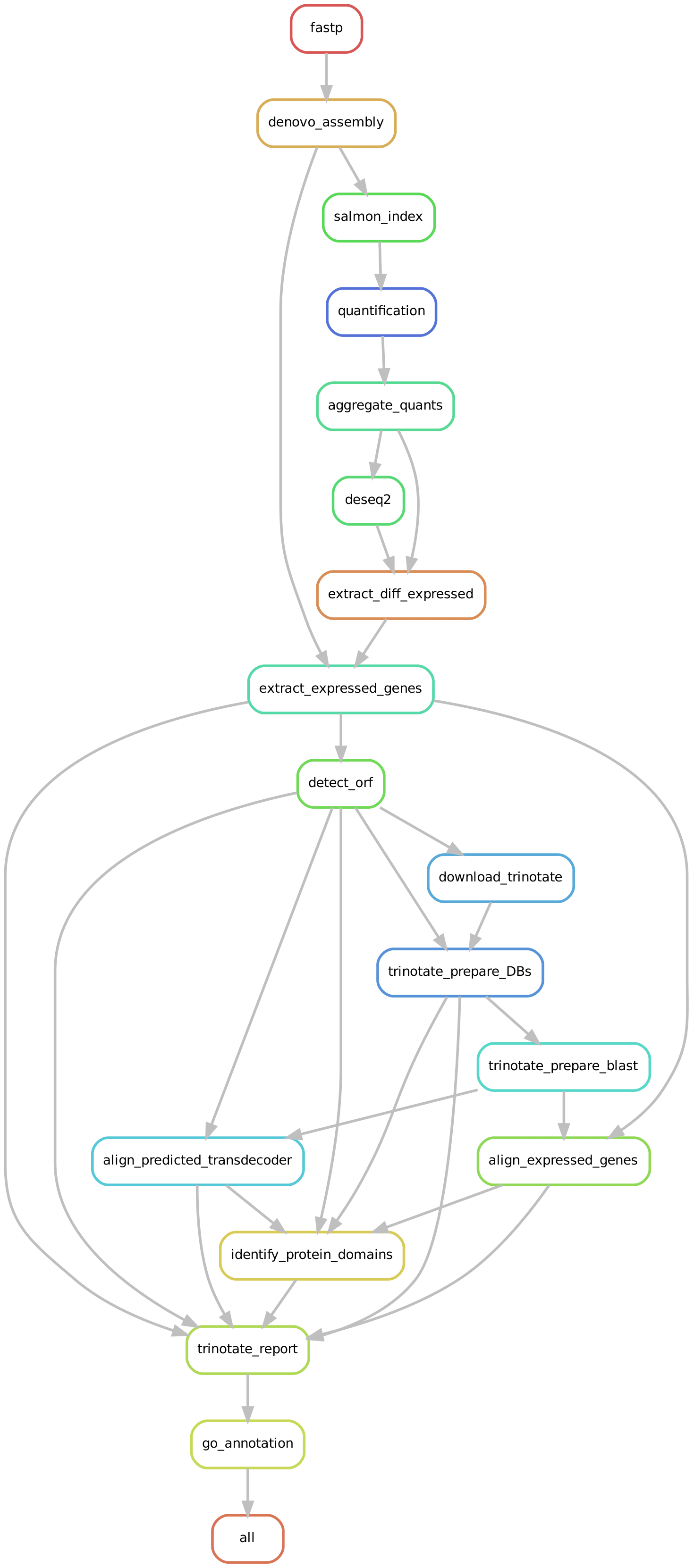
- This is a simple Snakemake workflow to perform de novo transcriptome assembly, DGE, and annotation.
- The workfow currently annotates the highly differential expressed genes with PVal of 0.005 and log_fold_change of 1.
conda env create -f environment.yml
conda activate rnaseq
- This workflow is meant to be easy to run, fast to implement, so it's currently not supporting flexibile configs.
- If you need to change configuration and parameters you will need to edit the Snakefile.
- It's only supporting RNASeq paired-end gzipped samples (not interleaved).
- Error trimming: fastp
- De novo transcriptome assembly: rnaSpades
- Quantification: Salmon
- Differential gene expression: DESEQ2
- Annotation: Trinotate
- Create a working directory
- Change the
ROOT_DIRin theSnakefileto match your working directory. - Create a directory with the name
samplesinside the working directory. - Put your samples in the
samplesdirectory with the naming convection:- R1: <sample_name>_1.fastq.gz
- R2: <sample_name>_2.fastq.gz
- Copy paste the tab-delimited file samples.tsv in your workflow directory.
- Modify the
samples.tsvto match your samples. Columns as following (sample_type, sample_name, R1_path, R2_path).
You may use the following bash commands to update the Snakefile and samples.tsv files.
curr=$(pwd)/workflow/
sed -i "s/REPLACE_ABSOLUTE_PATH/${curr//\//\\/}/g" workflow/samples.tsv
sed -i "s/REPLACE_ROOT_DIR/${curr//\//\\/}/g" Snakefile