Multi-omics disease sub-type specific drug repositioning aided with expression signatures from ConnectivityMap.
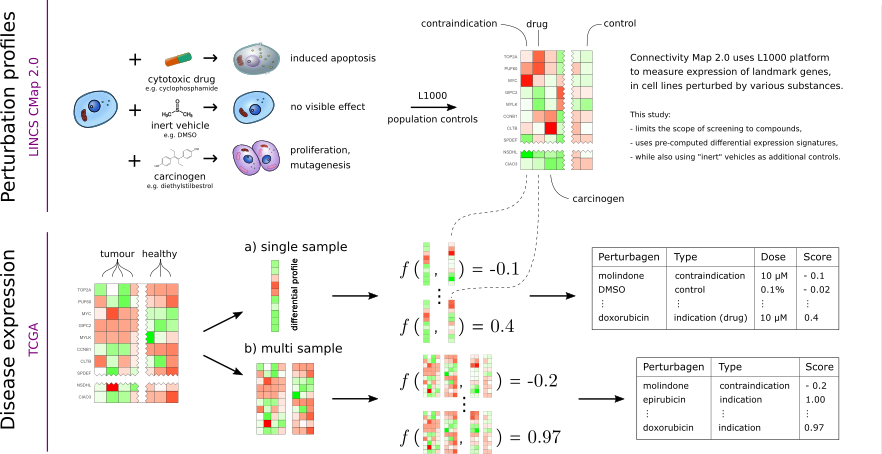


Attempts to guide the selection of drug candidates with machine learning has increased in recent years. One of the popular approaches is so-called "guilt-by-association" (GBA), using drug-drug and disease-disease similarity to guide the drug candidates selection. Comparison of genes expression profiles (perturbation profiles) after treatment with the candidate substances (perturbagenes) is a well-established approach to generate compound similarity maps for GBA methods. Large-scale projects, such as The Connectivity Map, offer ways of systematic perturbagene screening.
It was proposed that the perturbation profiles may be also used to find drug candidates by matching the profiles against differential expression profiles of diseases (being a simpler alternative to advanced machine-learning methods). This approach is referred to as pattern- or profile-matching and in the simplest setting corresponds to searching for anticorrelation of drug-disease profiles.
Previous studies demonstrated the merits of multi-omics disease stratification, evaluating the predictive ability of novel clusters for cancer patients survival or analyzing the functional enrichment in the clusters.
In this work, perturbagen-disease profile matching is applied to diseases and disease sub-types selected by multiple multi-omics stratification methods, in order to prioritize new drug repositioning candidates.
Multiple perturbation profile-disease expression matching methods (scoring functions) are evaluated, and then applied to cancer cohorts having enough data.
Gene-set enrichment (GSE) based methods are hypothesized to provide overall benefit by incorporation of additional biological information and availability of stringent significance estimates.
Finally, a hypothesis that scoring functions may be used to recognize stratifications based on meaningful molecular clustering, using only drug indications-contraindications classification performance is proposed.
Cancer data from The Cancer Genome Atlas are used, with an extensive case study on breast carcinoma (BRCA) cohort, limited validation with prostate (PRAD) and skin (STAD) adenocarcinomas, and a pan-cancer analysis.
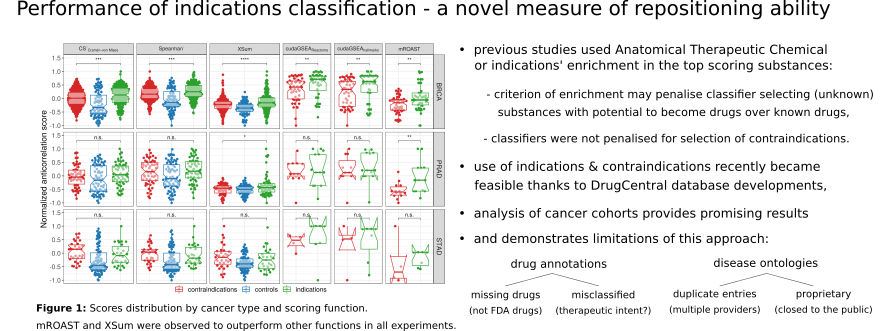
Indications-contraindications classification performance is used for scoring function evaluation. The evaluation was performed on 16 scoring functions of which six were proposed in previous works. Six scoring functions are chosen and applied in further analyses.
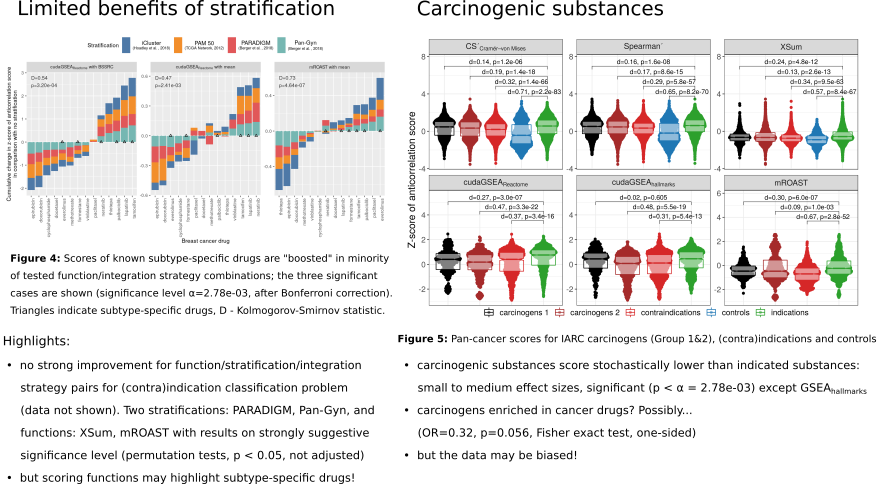
Performance of the scoring functions is compared using four previously published stratifications of breast cancer (including three based on multi-omics data): PAM50, iCluster, PARADIGM and Pan-Gyn.
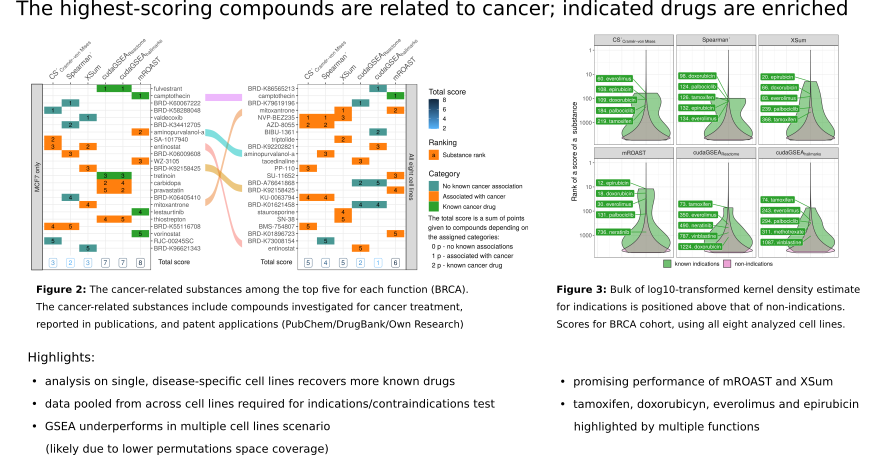
- The ability of profile-matching approaches to recover known drugs (as previously reported) is confirmed.
- A few previously unreported breast-cancer drug candidates are highlighted.
- The advantages and disadvantages of proposed indications-contraindications classification use.
- Multiple cancer drugs are noted to be known carcinogenic substances
- GSE-based methods require large numbers of samples, high-performance computing facilities and may not increase the chances of drug recovery in certain circumstances.
While the results obtained with meaningful stratifications do not always perform better than random permutations, limited benefit of stratification is observed for the drug recovery performance, with promising results from XSum and mROAST scoring functions.
Despite no definite evidence for the superiority of multi-omics stratifications use for classification of drug indications-contraindications, two multi-omics stratifications are highlighted as performing better than others: PARADIGM and Pan-Gyn.
Recommended packages for Ubuntu can be installed with:
bash ubuntu_setup.shPython in version 3.7 is recommended (minimum CPython 3.6). To install the required Python packages run:
pip3 install -r requirements.txtR in version 3.5.1 is required; the dependencies can be easily installed with:
Rscript install.RFinally, two major third-party applications (GSEA from Broad Institute, and custom fork of cudaGSEA) can be installed with:
cd thirdparty
bash download.shcudaGSEA needs to be compiled with:
./thirdparty/cudaGSEA/cudaGSEA/src/compileLimited number of unit tests is included and can be run to verify corresponding application fragments and integrity of the installation with:
./run_tests.shEach of the data sources has corresponding subdirectory (in data directory)
containing download.sh script, which will download the required data.
For example, to download TCGA data use:
bash data/tcga/download.shIf you wish to reproduce only part of the findings, you may want to download only required sources due to large file sizes.
The cells, RNA, DNA and histone pictograms are derivative works based on graphics from Reactome Icon Library (licensed under Creative Commons Attribution 4.0 International License).
The code in this repository was written as a part of MRes research project at Imperial College London. The research was conducted under the supervision of Dr Paul-Michael Agapow.
TBD