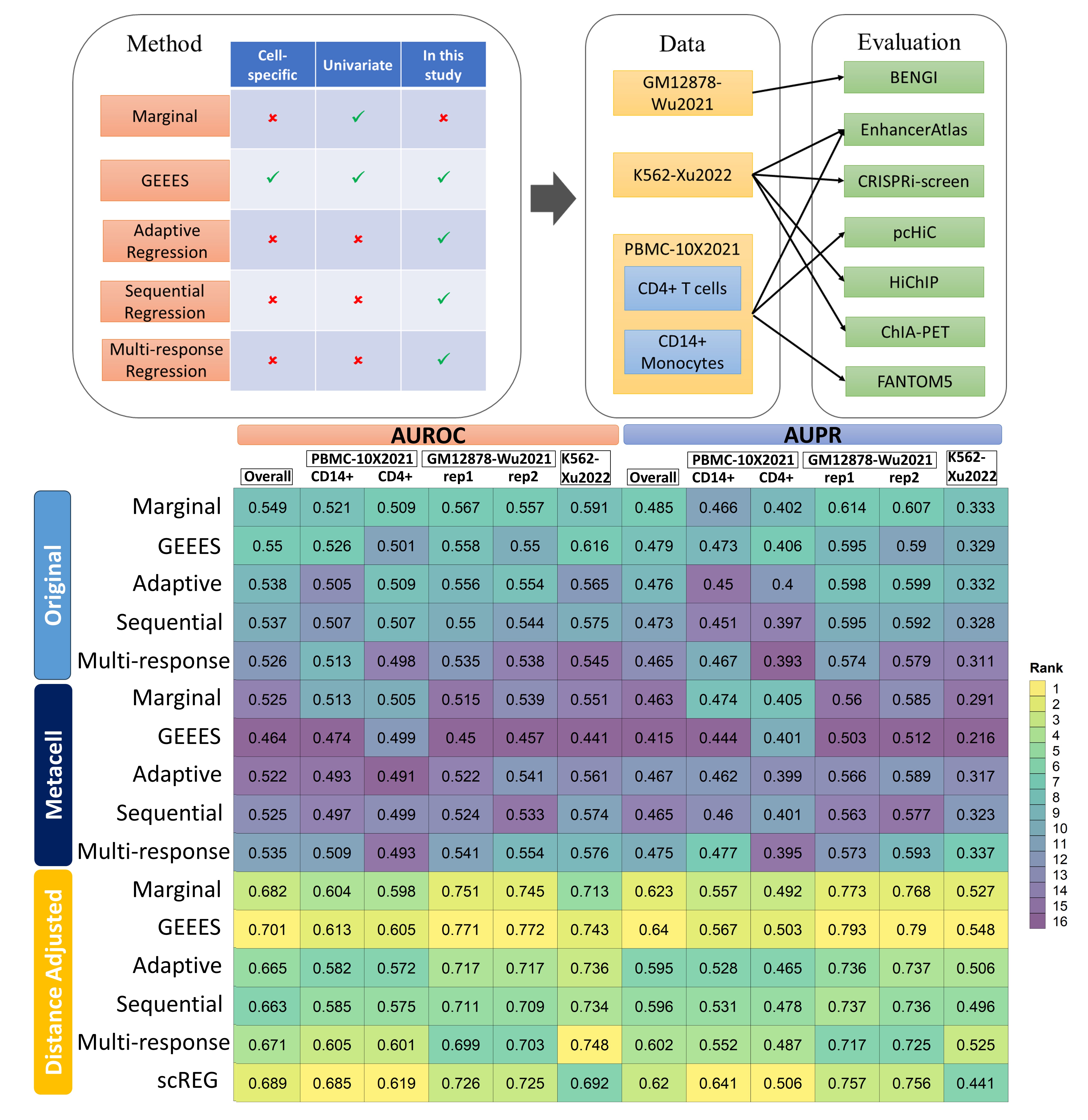
GEEES is a novelly proposed approach for inferring cell-specific Gene-EnhancEr IntEractions from Multi-modal Single Cell Data with transcriptome and chromatin accessibility profiles. GEEES estimates gene-enhancer associations at the single cell level by considering a cell neighbourhood defined by both the expression of the gene and the accessibility of the enhancer in the gene-enhancer pair.

## install.packages("devtools")
devtools::install_github("Shuyang12138/GEEES")If the installation fails, make sure you can install the following R packages:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("EnsDb.Hsapiens.v86")
BiocManager::install("biovizBase")
BiocManager::install("Seurat")
BiocManager::install("SeuratObject")
BiocManager::install("IRanges")
BiocManager::install("Signac")
BiocManager::install("GenomicRanges")
BiocManager::install("GenomeInfoDb")
BiocManager::install("MatrixGenerics")
install.packages("pbapply")
devtools::install_github("cran/remMap")library(Signac)
library(EnsDb.Hsapiens.v86)
library(GEEES)
# data preparation
K562_mini_obj <- CreateSeuratObject(counts = K562_mini$SCT)
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevelsStyle(annotations) <- 'UCSC'
genome(annotations) <- "hg38"
chrom_assay <- CreateChromatinAssay(
counts = K562_mini$ATAC,
genome = 'hg38',
annotation = annotations
)
K562_mini_obj[["ATAC"]] <- chrom_assay
# Results without distance adjustment
GEEES_mini <- NetworkInCell(K562_mini_obj,peak.assay = "ATAC",expression.assay="RNA",peak.slot = "counts",expression.slot = "counts",genes.use = NULL,cl = NULL,num=30)
GEEES_mini_aggregated <- generate_pair(GEEES_mini)
set.seed(2023)
adaptive_mini <- adaptive.regress.stabs(coaccess.adapt.data.K562,K562_mini_obj,peak.assay = "ATAC",expression.assay="RNA",peak.slot = "counts",expression.slot = "counts",PFER = 0.8,q = 3)
sequential_mini <- sequential.regression.stabs(coaccess.adapt.data.K562,K562_mini_obj,peak.assay = "ATAC",expression.assay="RNA",peak.slot = "counts",expression.slot = "counts",PFER = 0.8,q = 3)
multiResponse_mini <- remMap.regression.stabs(coaccess.adapt.data.K562,K562_mini_obj,peak.assay = "ATAC",expression.assay="RNA",peak.slot = "counts",expression.slot = "counts",PFER = 0.8,q = 2)
# Results with distance adjustment
gene.coords <- CollapseToLongestTranscript(annotations)
gene.coords <- data.frame(gene.coords)
rownames(gene.coords) <- gene.coords$gene_name
GEEES_mini_aggregated <- Distance_adjust(GEEES_mini_aggregated,gene.coords)
adaptive_mini <- Distance_adjust(adaptive_mini,gene.coords)
sequential_mini <- Distance_adjust(sequential_mini,gene.coords)
multiResponse_mini <- Distance_adjust(multiResponse_mini,gene.coords)GEEES is benchmarked against the state-of-the-art methods and a number of multivariate regression approaches we devised on a diverse set of multi-modal single cell datasets using a wide variaty of gold standard gene-enhancer interaction datasets. The datasets involved and summarized result table is shown as below.