This document is an exploration/set of notes on how we might adapt Nextflow or Snakefllow workflows into the OSCAR environment. It'll cover:
- Quick-start example of running
nf-core/nanoseqpipeline - Quick-start example of writing a custom Nanopore QC/metagenomics pipeline
nanopore_qc.nfand control flow basics - Configuring/customizing Nextflow (logging locations, volume mounting)
- Nextflow advanced control-flow
Channel
Motivation: nf-core/nanoseq is a general purpose analysis pipeline for Nanopore DNA/RNA data "that can be used to
perform basecalling, demultiplexing, QC, alignment, and downstream analysis."; we want to figure out how to run it in its
"minimal viable/hello world" version, how to set up its input sample sheet and run a simple reads QC task on our custom reads.
- Pull down the
compbiocore/workflows_on_OSCARproject and install the package on your OSCAR environment.
git clone https://github.com/compbiocore/workflows_on_OSCAR.git
# follow the interactive screen and instructions
bash ~/workflows_on_OSCAR/install_me/install.sh && source ~/.bashrc
- Make your samplesheet
samplesheet.csv
You can download an E.coli example read here: https://figshare.com/articles/dataset/Ecoli_K12_MG1655_R10_3_HAC/11823087
and scp it into your home directory ($HOME/test_run/ecolk12mg1655_R10_3_guppy_345_HAC.fastq.gz).
Please replace $HOME with your home directorypath in the samplesheet below:
You can download an E.coli example read here: https://figshare.com/articles/dataset/Ecoli_K12_MG1655_R10_3_HAC/11823087
and scp it into your home directory ($HOME/test_run/ecolk12mg1655_R10_3_guppy_345_HAC.fastq.gz).
Please replace $HOME with your home directory path in the samplesheet below:
cd $HOME/test_run
# make the samplesheet
echo group,replicate,barcode,input_file,fasta,gtf > samplesheet.csv
echo WT,1,,$HOME/test_run/ecolk12mg1655_R10_3_guppy_345_HAC.fastq.gz,, >> samplesheet.csv
- Run on OSCAR
nextflow_start
# skip every step except the QC of the reads
nextflow run nf-core/nanoseq --input $HOME/test_run/samplesheet.csv --protocol DNA --skip_basecalling --skip_demultiplexing --skip_quantification
- See Your Result:
When everything is done, you should see something like below:

All of the results of sub-tasks should be in the directories referenced above in the work directory of where you ran the
original nextflow run command , e.g., if you want to look at the Nanopolot
QC statistics of the reads,
# referenced as in [83/61b87c] process > NFCORE_NANOSEQ:NANOSEQ:QCFASTQ_NANOPLOT_FASTQC:NANOPLOT (WT_R1)
ls -la work/48/36d7e0d987eb42770d52dbf4bb96a9
output:
. .command.err .command.run ecolk12mg1655_R10_3_guppy_345_HAC.fastq.gz WT_R1_1_fastqc.html WT_R1_2.fastq.gz
.. .command.log .command.sh .exitcode WT_R1_1_fastqc.zip
.command.begin .command.out .command.trace versions.yml WT_R1_1.fastq.gz
You can scp the actual html output of FastQC WT_R1_1_fastqc.html in the output directory into your local machine and open it, it should like below:

You can also inspect the various execution files .command.run what command is run inside the container:
#!/bin/bash -euo pipefail
[ ! -f WT_R1_1.fastq.gz ] && ln -s ecolk12mg1655_R10_3_guppy_345_HAC.fastq.gz WT_R1_1.fastq.gz
[ ! -f WT_R1_2.fastq.gz ] && ln -s null WT_R1_2.fastq.gz
fastqc --threads 6 WT_R1_1.fastq.gz WT_R1_2.fastq.gz
also .command.run for the command submitted to the job scheduler; .command.err and .command.log for the std-err
and std-out respectively.
Motivation: we might not always rely on nf-core's workflows and may want to write our own workflows. Here we are going
to write a simple workflow that does QC on Nanopore reads from scratch.
- Write the first part of the Nextflow pipeline in your favorite editor (just in the end have to have it as an .nf file on your OSCAR environment):
Our pipeline will take either Fastq reads --read or the sequencing summary file from MinION basecaller --sequencing_summary
or the raw files from the MinION sequencer --fast5. We check for this in the follow text below.
#! /usr/bin/env nextflow
nextflow.enable.dsl=2
if (!params.read && !params.sequencing_summary && !params.fast5) {
error "Error: at least one input format (--read, --sequencing_summary, --fast5) must be enabled."
}
Just a quick note that Nextflow's scripting language is based off Groovy which is very similar to the conditional flow of Java, Python and C#.
- Write the first task in our Nextflow pipeline:
process guppy {
container 'cowmoo/pycoqc:latest'
input:
file fast5s
output:
path "guppy_out/*.fastq", emit: reads
path 'guppy_out/sequencing_summary.txt', emit: sequencing_summary
"""
/ont-guppy-cpu/bin/guppy_basecaller -i ${fast5s} -s guppy_out -c dna_r9.4.1_450bps_hac.cfg --num_callers 8 --cpu_threads_per_caller 1
"""
}
The process is a sub-task of the pipeline. Here the guppy process/sub-task is a task we will run the Guppy basecaller
to generate fastq files from the raw fast5 files from Nanopore sequencers.
The container is the Docker or Singularity container we will use to run the job in. Container tutorial is out of the scope
of this tutorial. See Building Docker documentation for more details.
comwoo/pycoqc:latest is a dedicated pre-built container on Dockerhub for this tutorial.
input and output are pretty self-explainatory. Note how the inputs fast5s are then integrated into guppy_basecaller command.
And how outputs reads and sequencing_summary.txt are extracted as outputs respectively as a set of .fastq files and single text file.
- Write out the rest of the
processof the Nanopore QC workflow:
process concat_reads {
container 'cowmoo/pycoqc:latest'
input:
path reads
output:
file "reads.fastq", emit: read
"""
cat ${reads} > reads.fastq
"""
}
process pycoQC {
container 'cowmoo/pycoqc:latest'
input:
file reads_summary
output:
path "pycoQC_output.html"
"""
pycoQC -f ${reads_summary} -o pycoQC_output.html
"""
}
process kraken {
container 'cowmoo/pycoqc:latest'
containerOptions '-v /Users/paulcao/Downloads/minikraken2_v2_8GB_201904_UPDATE:/db'
input:
file reads
output:
path "out.txt"
"""
/kraken2/kraken2 -db /db ${reads} --gzip-compressed --output out.txt --report report.txt
"""
}
process nanoPlot {
container 'cowmoo/pycoqc:latest'
input:
file input
output:
path "summary-plots-log-transformed/*"
script:
if (params.reads_summary) {
"""
NanoPlot --summary ${input} --loglength -o summary-plots-log-transformed
"""
}
else if (params.read) {
"""
NanoPlot --fastq ${input} --loglength -o summary-plots-log-transformed
"""
}
}
- Define the main flow block fo the pipeline:
This section will go in sequential order, the code of the main workflow and in English what the current code block is doing.
workflow {
/* run guppy if fast5 directory is supplied */
if (params.fast5s) {
fast5 = file(params.fast5)
guppy(fast5)
}
...
}
The above block checks if the the parameter --fast5 is supplied, if so, it'll invoke the guppy process to try to base
call the fast5 files to get resulting fastq files.
/* run taxonomical identification, either by guppy's called reads or user-suplied reads */
read = null
if (params.fast5s || params.read) {
read = (params.fast5s) ? concat_reads(guppy.out.reads.collect()) : file(params.read)
kraken(read)
}
This above block attempts to taxonomically identify what the reads are using kraken. If the reads was
supplied by --read, Kraken will be run on those reads. However if the user supplied a --fast5 argument, the pipeline
will take the previously run guppy process and also concat all of the reads into a single fastq file via concat_read process; and
then run the Kraken on it.
/* run reads QC, either by the raw reads or user-supplied reads_summary.txt */
if (params.reads_summary) {
reads_summary = file(reads_summary)
nanoPlot(reads_summary)
pycoQC(reads_summary)
} else if (params.fast5s) {
reads_summary = file(guppy.out.reads_summary.collect())
nanoPlot(reads_summary)
pycoQC(reads_summary)
} else if (read) {
nanoPlot(read)
}
This above block attempts to run QC on the user's supplied inputs. Some of the QC tools can only be run on
reads_summary.txt supplied by MinION basecallers; some can be run on either FastQ reads or reads_summary.txt.
So if --reads_summary is supplied, we run both nanoPlot and pycoQC using the text file as the input.
However if --fast5 is supplied, we then retrieve the reads_summary.txt gathered from the previously run guppy process;
and run nanoPlot and pycoQC with that input.
Finally if only --read is supplied, then we run only nanoPlot as the only QC tool that can process the fastq files.
See here: workflows/nanoqc.nf
nextflow run nanoqc.nf --reads_summary input/reads_summary.txt
nextflow run nanoqc.nf --fast5 input/fast5_drectory
nextflow run nanoqc.nf --read input/test_read.fastq.gz
By default Nextflow outputs are stored in /work directories; under automatically generated uuid directories.
The following example demonstrates how to generate a more human readable output directory organized by sample id; (or any permutations using input and output parameters).
See particularly publishDir parameter; if the workflow is run with --outdir $HOME/workflow_out and given a sample-sheet
with a sample_id of WT.
process pycoQC {
debug true
container 'pycoqc'
publishDir "$params.outdir/$sample_id"
input:
tuple val(sample_id), file(summary), file(reads), file(fast5)
output:
path "pycoQC_output.html"
script:
if (summary)
"""
pycoQC -f ${summary} -o pycoQC_output.html
"""
}
Then for this example, Nextflow will then output to the directory: $HOME/workflow_out/WT/pycoQC_output.html
For some job, we may have to mount to external databases (e.g., BLAST databases, gene annotation or pathway databases).
We can do this using the containerOption parameter. The following example demonstrate how to mount a Kraken database path
as a /db/ path inside a container; and using it to run a Kraken classification job.
process kraken {
debug true
container 'pycoqc'
publishDir "$params.outdir/$sample_id/kraken"
containerOptions '--bind /Users/test_user/minikraken2_v2_8GB_201904_UPDATE:/db'
input:
tuple val(sample_id), file(summary), file(reads), file(fast5)
output:
path "out.txt"
"""
/kraken2/kraken2 -db /db ${reads} --gzip-compressed --output out.txt --report report.txt
"""
}
process kraken {
debug true
container 'pycoqc'
publishDir "$params.outdir/$sample_id/kraken"
containerOptions '-v /Users/test_user/minikraken2_v2_8GB_201904_UPDATE:/db'
input:
tuple val(sample_id), file(summary), file(reads), file(fast5)
output:
path "out.txt"
"""
/kraken2/kraken2 -db /db ${reads} --gzip-compressed --output out.txt --report report.txt
"""
}
It is important to find the logs of failed runs and also customize the logs for either debugging purposes or to support/debug yours or other users' failed runs.
There are several ways to do this, (1) by using the Nextflow CLI to query and display the workflow's logs. (2) by using a custom logging template, (3) by running a Nextflow with a report.
# show all recent ran nextflow query for the current user
nextflow log
output:
TIMESTAMP DURATION RUN NAME STATUS REVISION ID SESSION ID COMMAND
2022-11-16 13:37:51 - clever_brahmagupta - 1e60482a2c e3372fc2-f6d8-4c14-8032-73d02ae6c634 nextflow run nf-core/nanoseq --input test_run/samplesheets.csv --protocol DNA --skip_basecalling --skip_demultiplexing --skip_quantification
...
(the RUN_NAME are automatically generated; however you can add your own label to your workflow in your nextflow run with
a ---name)
You can then query the entire working directory and the stdout and std-err for each proceses of the workflow as follows:
nextflow log clever_brahmagupta -f workdir,name,exit,status
output:
/gpfs/home/me/work/04/c911cff4dd97f43cd1537371e533bf NFCORE_NANOSEQ:NANOSEQ:INPUT_CHECK:SAMPLESHEET_CHECK (samplesheet.csv) 0 COMPLETED
/gpfs/home/me/work/83/61b87c6b30c0ed95f17d11947c1293 NFCORE_NANOSEQ:NANOSEQ:QCFASTQ_NANOPLOT_FASTQC:NANOPLOT (WT_R1) 0 COMPLETED
/gpfs/home/me/work/48/36d7e0d987eb42770d52dbf4bb96a9 NFCORE_NANOSEQ:NANOSEQ:QCFASTQ_NANOPLOT_FASTQC:FASTQC (WT_R1) 0 COMPLETED
/gpfs/home/me/work/a3/afb0a76cddbc0bed0be89ae0ddc4ac NFCORE_NANOSEQ:NANOSEQ:CUSTOM_DUMPSOFTWAREVERSIONS (1) 0 COMPLETED
/gpfs/home/me/work/86/3088143ac9c4d4878cb954e5795f7e NFCORE_NANOSEQ:NANOSEQ:MULTIQC (1) 0 COMPLETED
You could write a Markdown or HTML template that would summarize every single process in a workflow into a single file. e.g.,
template.md:
## $name
script:
$script
exist status: $exit
task status: $status
task folder: $folder
write a log of every process in a workflow:
nextflow log goofy_kilby -t template.md > execution-report.md
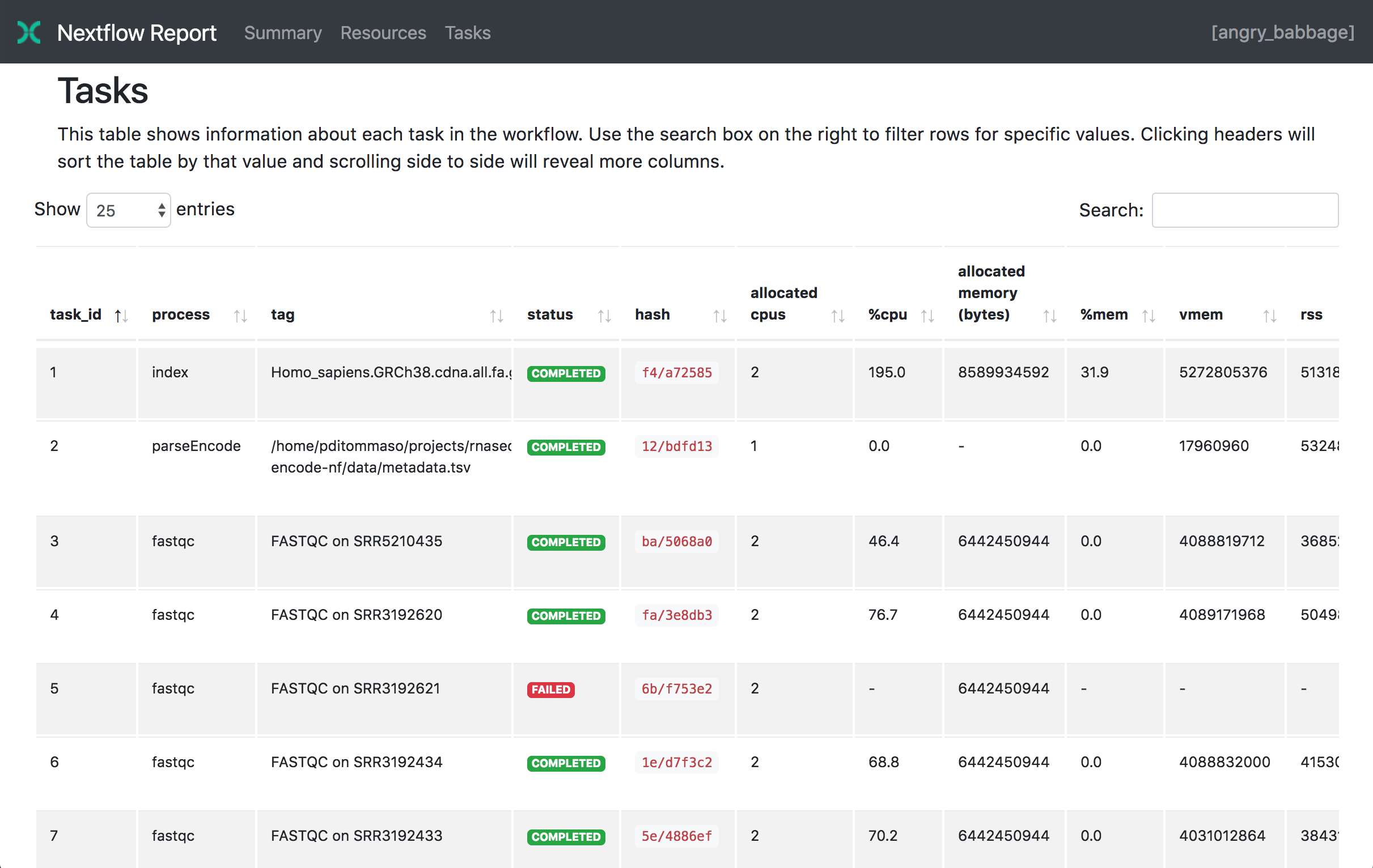
You could run a Nextflow workflow --with-report parameter; and the resulting workflow
will output a single HTML file containing a breakdown of every single process.
nextflow run <pipeline name> -with-report [file name]
output:
Motivation: Basic control-flows of if and else seems to be sufficient for most use cases. But what if you have a
complex sample sheet or an complex intermediate data structure in your workflow - that requires different processes.
(e.g., a samplesheet containing a mixed rows of long reads or short reads; an annotation pipeline that needs to process a mix of both prokaryotes or eukaryotes organisms).
Nextflow provides a mechanism to do this called Channel where you can iterate/filter/map over each item in a data
structure (similar to Python lambda expressions); and fork each item to the right sub-workflow.
Samplesheet:
Suppose there is a WT_1 and a A1 sample; respectively having a fastq file or a fast5 directory associated with it. Therefore,
a QC workflow taking this samplesheet will have to create a Channel from the sample sheet CSV; and filter for the samples
with a FAST5 input and run base-calling on it first; and another filter for FASTQ input to run QC directly.
sample_id,read_summary,reads,fast5
WT_1,/users/test/inputs/sequencing_summary.txt,/users/pcao5/inputs/ecolk12mg1655_R10_3_guppy_345_HAC.fastq.gz,
A1,,,/users/test/inputs/fast5_directory
workflow {
/* first filter in the samplesheet, rows where fast5 is specified; run Guppy */
Channel.fromPath(params.samplesheet).splitCsv(header:true)
.filter(row -> row.fast5)
.set { fast5_ch }
BASE_CALL (fast5_ch).set { base_called_ch }
/* then filter for in the samplesheet, rows where fastq or sequencing_summary.txt is specified */
Channel.fromPath(params.samplesheet).splitCsv(header:true)
.filter(row -> (row.summary || row.reads) && !row.fast5)
.map { get_sample_info(it) }.set { fastq_ch }
/* now both fastq_ch and base_called_ch have fastq files, combine both channels
and process fast1 files*/
PROCESS_SAMPLE (fastq_ch.concat(base_called_ch))
emit:
PROCESS_SAMPLE.out
}
// Function to resolve files and verify files exist
def get_sample_info(LinkedHashMap sample) {
summary = sample.read_summary ? file(sample.read_summary, checkIfExists: true) : null
reads = sample.reads ? file (sample.reads, checkIfExists: true) : null
fast5 = sample.fast5 ? file(sample.fast5, checkIfExists: true) : null
return [ sample.sample_id, summary, reads, fast5 ]
}
See here: workflows/nanoqc.nf
nextflow run nanoqc_samplesheet.nf --samplesheet samplesheet.csv