
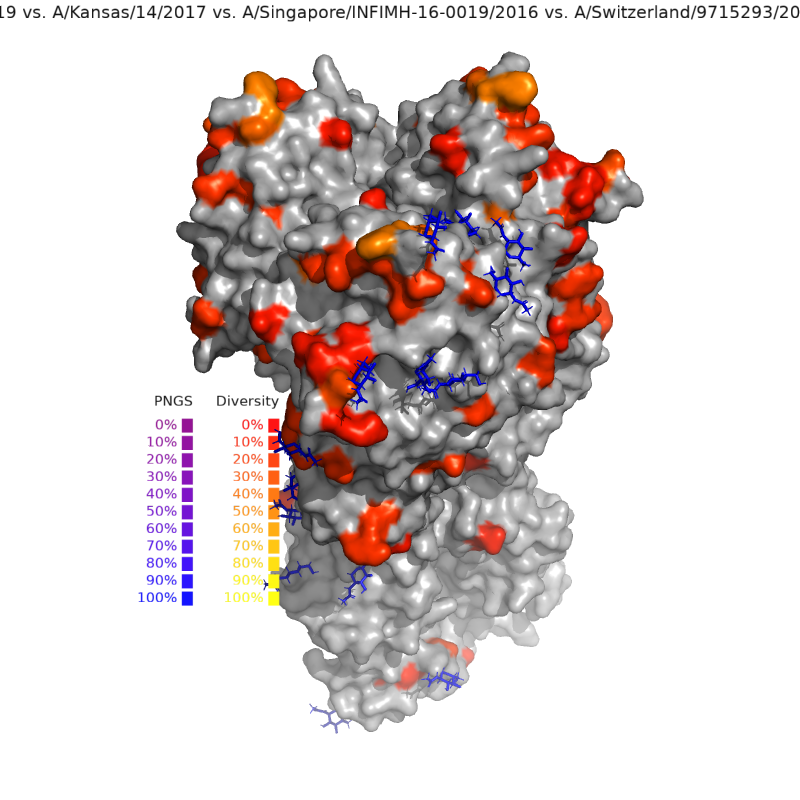
Flu Strain Compare generates visualizations of mutations between pairs of HA sequences. make_comparison_figure.py takes two HA sequences as input and outputs a figure highlighting amino acid and PNGS changes on a representative HA crystal structure. At present, H1pdm and H3 strains are supported.
This utility runs on Docker, Singularity, or natively with PyMOL installed as a Python module.
You can use the following command to build the container:
docker build ./ -t flu_strain_compare
Build Docker container above. This may need to be done locally if your HPC system doesn't allow Docker.
# load module, if in HPC environment
module load singularity
singularity build ubuntu-pymol-biopython_latest.sif docker-daemon://flu_strain_compare:latest
Install PyMOL as a Python library.
# Optional, if using a virtual environment.
python -m venv venv
source venv/bin/activate
# Install python dependencies.
pip install Bio pandas
pip install scipy
# Install PyMOL as a library.
PYMOL_VERSION=2.5.0
wget --no-verbose https://github.com/schrodinger/pymol-open-source/archive/refs/tags/v${PYMOL_VERSION}.tar.gz
tar xfz v2.5.0.tar.gz
cd pymol-open-source-2.5.0
git clone --depth=1 https://github.com/rcsb/mmtf-cpp.git
cd mmtf-cpp
git pull
cd ..
cp -r mmtf-cpp/include/mmtf* include/
python3 setup.py install
- Install python dependencies.
pip install Bio pandas
pip install scipy
-
Navigate to https://github.com/schrodinger/pymol-open-source/archive/refs/tags/v2.5.0.tar.gz in a browser and save the file.
-
Uncompress v2.5.0.tar.gz using a compression utility such as 7-Zip.
-
Copy
mmtf-cpp/include/mmtf/andmmtf-cpp/include/mmtf.hppto theinclude/directory inpymol-open-source-v2.5.0(the directory in the decompressed archive above). -
Install PyMOL as a library.
python3 setup.py install
The configuration file specified in the command line (see Running below) serves as input for the make_comparison_figure.py script.
seq_file: Name of fasta-formatted file that contains full-length amino acid HA sequences. File must be indatadirectory.q1_id: Sequence ID of the first query strain. The sequence id is the first word in the fasta header of the desired sequence.q2_id: Sequence ID of the second query strain.seq_lineage: Lineage of the query strains."H1"or"H3".numbering_scheme: What numbering scheme do you want to use for mutation identification? For H1 sequences, you can chooseH1pdm,H3, orH1_1933. For H3 sequences, onlyH3numbering is available.- ``: What numbering scheme do you want to use for mutation identification? For H1 sequences, you can choose
H1pdm, `H3`, or `H1_1933`. For H3 sequences, only `H3` numbering is available. reference_mode: Use the reference strain for comparison (trueorfalse). Note: .png output is much more resource intensive than .pse.,export_files: Export .pse file, .png file, or both. Example:["PSE"],filter_sites: Filter out mutations and PNGS at the listed sites. Example:[92, 94, 104],reverse_filter_sites: Only include the listed sites, and exclude others. Note: reverse filter will override filter if both are non-empty. Example:[92]output_csv: Write CSV to standard output. Default:false(JSON).diversity_index:"shannon","gini-simpson","richness"(Default: Shannon).non-conservative_only: Only include non-conservative mutations to measure diversity.trueorfalse.
Notes
- Site numbers input in "filter_sites" and "reverse_filter_sites" refer to amino acid numbering in the final HA proteins, after signaling peptides have been removed, from the start of HA1. The output table includes both numbering schemes (with and without signaling peptides), for reference
Note: flu_strain_compare_path is an absolute path to the root of this repository.
export flu_strain_compare_path=/some/abs/path/flu_strain_compare
With your configuration file set up to your liking, run the container with the following commands:
docker run -v ${flu_strain_compare_path}:/app flu_strain_compare python3 src/<SCRIPT NAME>.py configuration/config.json
singularity exec --bind ${flu_strain_compare_path}:/app ubuntu-pymol-biopython_latest.sif python3 src/<SCRIPT NAME>.py configuration/config.json
python src/<SCRIPT NAME>.py configuration/config.json
<SCRIPT NAME> should be make_comparison_figure
Example:
# from repo root
singularity exec --bind /home/youruser/flu_strain_compare:/app ubuntu_pymol_biopython.sif python3 src/make_comparison_figure.py configuration/config.json
Outputs will be written to the figures directory of the repository.
If you wish the csv with additional information to be printed out, include
pytest
- 2021-2022 southern hemisphere cell-based recommendation (name =
A/Darwin/6/2021, id =EPI1885402) - 2021-2022 northern hemisphere Flublok (name =
A/Tasmania/503/2020, id =EPI1752480) - 2021-2022 northern hemisphere cell-based recommendation (name =
A/Cambodia/e0826360/2020, id =EPI1843589) - 2020-2021 northern hemisphere Flublok (name =
A/Minnesota/41/2019, id =EPI1548699) - 2020-2021 northern hemisphere cell-based recommendation (name =
A/Hong Kong/45/2019, id =EPI1409001) - 2019-2020 cell-based recommendation (name =
A/Kansas/14/2017, id =EPI1174043) - 2018-2019 cell-based recommendation (name =
A/Singapore/INFIMH-16-0019/2016, id =EPI1106235) - 2016-2018 cell-based recommendation (name =
A/Hong Kong/4801/2014, id =EPI539576) - 2015-2016 cell-based recommendation (name =
A/Switzerland/9715293/2013, id =EPI530687) - 2013-2015 cell-based recommendation (name =
A/Victoria/361/2011, id =EPI349103)
- 2021-2022 northern hemisphere cell-based recommendation (name =
A/Wisconsin/588/2019, id =EPI1715168) - 2020-2021 northern hemisphere cell-based recommendation (name =
A/Hawaii/70/2019, id =EPI1669665) - 2019-2020 cell-based recommendation (name =
A/Brisbane/02/2018, id =EPI1212884) - 2017-2019 cell-based recommendation (name =
A/Michigan/45/2015, id =EPI699812 - 2009-2017 cell-based recommendation (name =
A/California/04/2009, id =EPI178457) - 2013-2014 example from Linderman et al. (name =
A/Colorado/3514/2013, id =EPI501723)
renumber_H1.pyandrenumber_H3.py: Re-index PDB files for use with Flu Strain Compare.print_pymol_objects.py: Print chains and selections in a .pse file to compare modifications to the model.