Home
All questions regarding this work can be directed to:
Gustavo A. Ramírez
email: ramirezg@westernu.edu
Twitter: @zombiephylotype
https://orcid.org/0000-0001-8122-4898
Here we will "demystify" the command line. Getting everyone on board with the proper software and data for a bioinformatics course is one of the most difficult part of teaching it. Fortunately for us, we will run our work using virtual "instances" access through binders. This will safe tons of time that is usually allocated to getting everyone's software (hardware dependent usually) to access and run commands from a terminal screen. The instance places us all individually on a small allocation of computer space/time that we access remotely use as our work space.
Just click the launch/binder icon and enter a fully-functional UNIX working environment! -fyi: it may take a few mins to load... When it does, scroll to the bottom and click on the $_Terminal icon to start a terminal window. This is your work environment :)
Listing files and directories
This command lists files and folders at the location where it is executed.
copy and paste (or type) the command below into your terminal screen. press enter.
ls
Unix core commands can do much more. They can be ran with "arguments" in the following way: command argument.
Try this:
ls -l
Your files are listed in alphabetical order...
Here:
ls -lS
your files are sorted by size!
for a full description of the possible arguments used with the core command "ls" try the following:
ls --help
... Quite an extensive list!... scroll up and down... don't shy...
To clear your screen simply type:
clear
and, of course, just out of curiosity, try:
clear --help
...huh...
and clear again:
clear
Take home here: Any command in a Unix system will work like this.... The more familiarized that you get with the "command argument" format, the easier time you'll have navigating through directories and executing Unix programs through the shell!
Let's explore Unix file structure!
A path is a literal address!
Knowing your RELATIVE or ABSOLUTE position in your computer is very important.
Note: Folders are called "directories", and each directory has its unique identifying address.
Not a truly new concept since the folder icons on your mac/pc GUI work the same way...
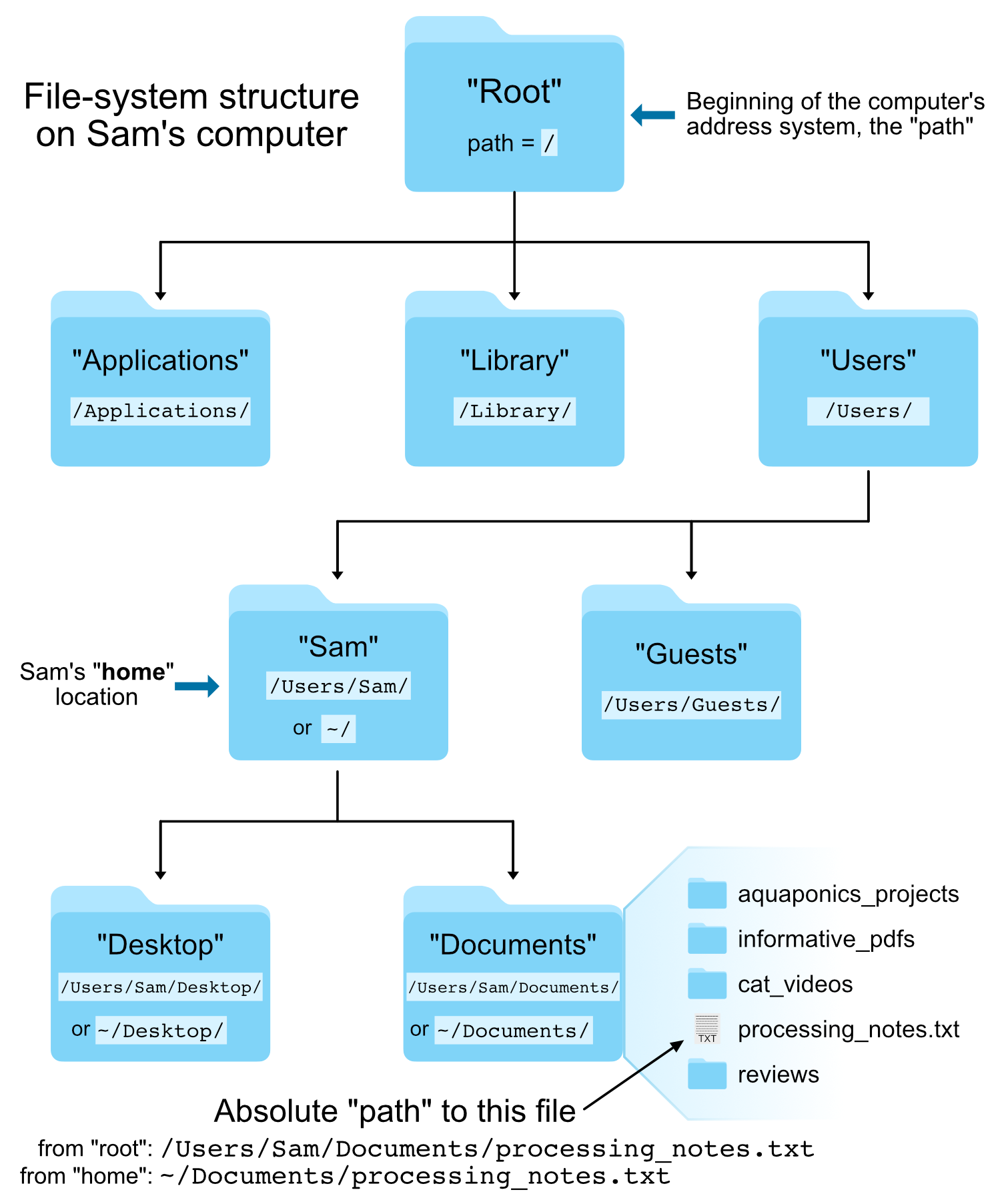
Image from @AstrobioMike
Let's check our address using the print working directory (pwd) command
pwd
The /home/jovyan folder is our home location. That is when we enter this terminal, we are dumped here!
Now, let's change directories (cd) to see what is in the "data" folder.
cd data
To list the contents of that folder use the ls command:
ls
and, indeed, confirm that we are now in the data directory (folder).
There are two additional directories here...
For fun, we can print our working directory (pwd) to confirm that we are in the data directory:
pwd
Now, let's move to the "ASVs" directory and lists its contents.
cd ASVs
ls
A single file (not a folder) is found here. Great... How do we get back "home"?
Two ways:
- directly return to your home directory
cd
pwd
- or do a step-wise return
cd ..
pwd
cd ..
pwd
Opening existing files and writing new ones
Let's jump directly to the ASVs directory as follows:
using the absolute path:
cd /home/jovyan/data/ASVs
or from your home path:
cd ~/data/ASVs
and list the file(s) again
ls
Some files can be quite large and/or not human readable.
Thus, when the content is unknown, to prevent time and computing resources from going into opening up a monster,
I have a sneak peak using the following commands: i) head, ii) tail, as follows:
head ASVs1_500.fa
tail ASVs1_500.fa
Head and tail list the first and last, respectively, 10 lines of the file. Our file prints more lines bc the text is "wrapped"; disregard...
Next we will checkout the files in the /genomes folder:
Can you navigate there alone? Try then drop down for hints!
One way is back one directory, then over to genomes:cd ..
cd genomes
Now, let's list (ls) the files in the genomes directory by "human-readable" size:
ls -lhs
and have a look at the first 10 lines in the 3.6M file called bin20.fasta.
head bin20.fasta
last 10 lines of the same file:
tail bin20.fasta
And finally, lets scroll through the entire file.
Note that this is a more complex operation that:
i) requires more memory/time
ii) may or many not be informative
iii) requires a "special exit" - lower case "q".
less bin20.fasta
to exit type "q" once.
If necessary, clear your screen.
Now, to create an empty file called "Hello.txt", use the following command:
touch Hello.txt
you can confirm the generation of this file and its empty state using the following:
ls
you should see it listed along with the other files
less Hello.txt
it should be empty... Exit with "q"!
Next, we will move this empty file one directory up:
mv Hello.txt ../
check this directory's content:
ls
Hello.txt is gone...
Let's move up a directory and check for it there!
Try it on your own, then drop down here
cd ..
ls
There it is!
Next, we will make a new directory called "New":
mkdir New
and we will use the mv command to move Hello.txt into this "New" folder:
mv Hello.txt New
Move in to the New folder and confirm that Hello.txt is there!
mv New
ls
There it is!
Happy Belly Bioinformatics: A fantastic resource!
Completion of the Unix crash course (~3-5h) prior to next class is highly recommended! https://astrobiomike.github.io/unix/