This is a bioinformatics analysis pipeline used for bulk RNA sequencing data developed at Zymo Research. This pipeline was adpated from community-developed nf-core/rnaseq pipeline version 1.4.2. Many changes were made to the original pipeline, some of which were similarly adopted by later versions of nf-core/rnaseq by coincidence. We also took some inspirations from versions 2.0.0 and 3.0.0. Zymo's modifications include, but are not limited to:
- Added differential gene expression analysis
- Added functional enrichment analysis
- Added/improved sample overview analysis, such as gene heatmap, MDS plot, etc.
- Added differential transcript usage analysis
- Added Salmon as an alternate way of read quantification
- Improved biotype QC analysis and rRNA reads detection
- Added ability to handle ERCC spike-ins
- Added several MultiQC plugins to visualize QC/analysis results
- Added modules to deal with UMIs within the Zymo-Seq SwitchFree 3′ mRNA Library Kit
- Compatibility to run on Aladdin Bioinformatics Platform
This pipeline is built using Nextflow, a bioinformatics workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It comes with docker containers making installation trivial and results highly reproducible.
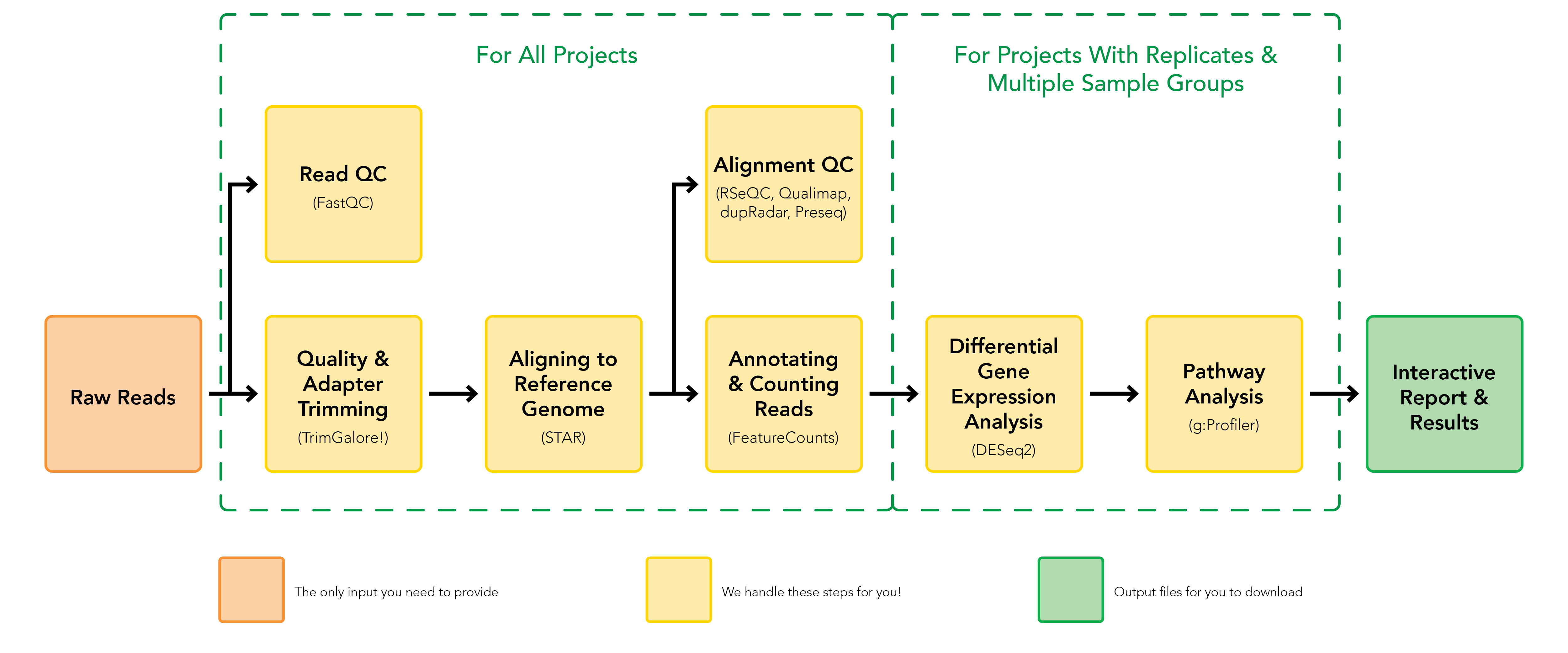
This pipeline processes raw data from FastQ inputs (FastQC, Trim Galore!), BBDuk, aligns the reads (STAR, generates gene or transcript read counts (featureCounts or Salmon), and performs extensive quality-control on the results (RSeQC, Qualimap, Picard, dupRadar, Preseq). This pipeline also conducts sample comparison analysis and differential gene expression analysis using DESeq2, and a functional enrichment analysis using g:Profiler. Optionally, the user could choose to run a differential transcript usage analysis modeled after this paper (DEXSeq, DRIMSeq, stageR). All QC and analysis results are visualized in a report using MultiQC).
- Nextflow version 20.07.1 or later
- Sufficient CPU and memory. Default is 8 threads and 60GB. Please modify nextflow.config file to fit your device.
- Docker if using
dockerprofile - Permissions to AWS S3 and Batch resources if using
awsbacthprofile
nextflow run Zymo-Research/aladdin-rnaseq \
--profile docker \
--genome 'Homo_sapiens[GRCh38]' \
--protocol Zymo_RiboFree_PROTv1 \
--design "<path to design CSV file>"- The parameters
--genome, and--protocolare required. Please refer to Usage Documentation for available options. - The parameter
--designis required. The design CSV file must have the following format.
group,sample,read_1,read_2
Control,Sample1,s3://mybucket/this_is_s1_R1.fastq.gz,s3://mybucket/this_is_s1_R2.fastq.gz
Control,Sample2,s3://mybucket/this_is_s2_R1.fastq.gz,s3://mybucket/this_is_s2_R2.fastq.gz
Experiment,Sample3,s3://mybucket/that_is_s3_R1.fastq.gz,
Experiment,Sample4,s3://mybucket/that_be_s4_R1.fastq.gz,
The header line must be present and cannot be changed. Sample labels and group names must contain only alphanumerical characters or underscores, and must start with a letter. Sample labels and group names also cannot start with "R1" or "R2", or include phrases that will be automatically filtered by MultiQC. A list of terms unsuitable for sample or group labels in this pipeline can be viewed in the MultiQC source code here. Full S3 paths of R1 and R2 FASTQ files should be provided. Mixing of paired-end and single-end data is allowed. If you do not wish to carry out comparisons of samples, i.e., DESeq2 and g:Profiler analysis, simply leave the Group column blank (but keep the comma).
nextflow run Zymo-Research/aladdin-rnaseq \
-profile awsbatch \
--genome 'Homo_sapiens[GRCh38]' \
--protocol Zymo_RiboFree_PROTv1 \
--design "<path to design CSV file>" \
-work-dir "<work dir on S3>" \
--awsqueue "<SQS ARN>" \
--outdir "<output dir on S3>" \
--name "<study name>"- The parameters
--genome, and--protocolare required. Please refer to Usage Documentation for available options. - The parameter
--designis required. See above for file format. - The parameters
--awsqueue,-work-dir, and--outdirare required when running on AWS Batch, the latter two must be directories on S3. - The option
--namewill add a title to the report.
There are many other options built in the pipeline to customize your run and handle specific situations, please refer to the Usage Documentation.
A sample report this pipeline produces can be found here.
A documentation explaining how to understand the report can be found here.
A Materials and Methods document is available to help you write your manuscript if you used this pipeline in your research.