When doing single-cell RNA sequencing work, we firstly do clustering by community detection. Then we match the clusters to major cell types, which forms a cluster tree. Following cluster tree represents Allen Human Brain Atlas:
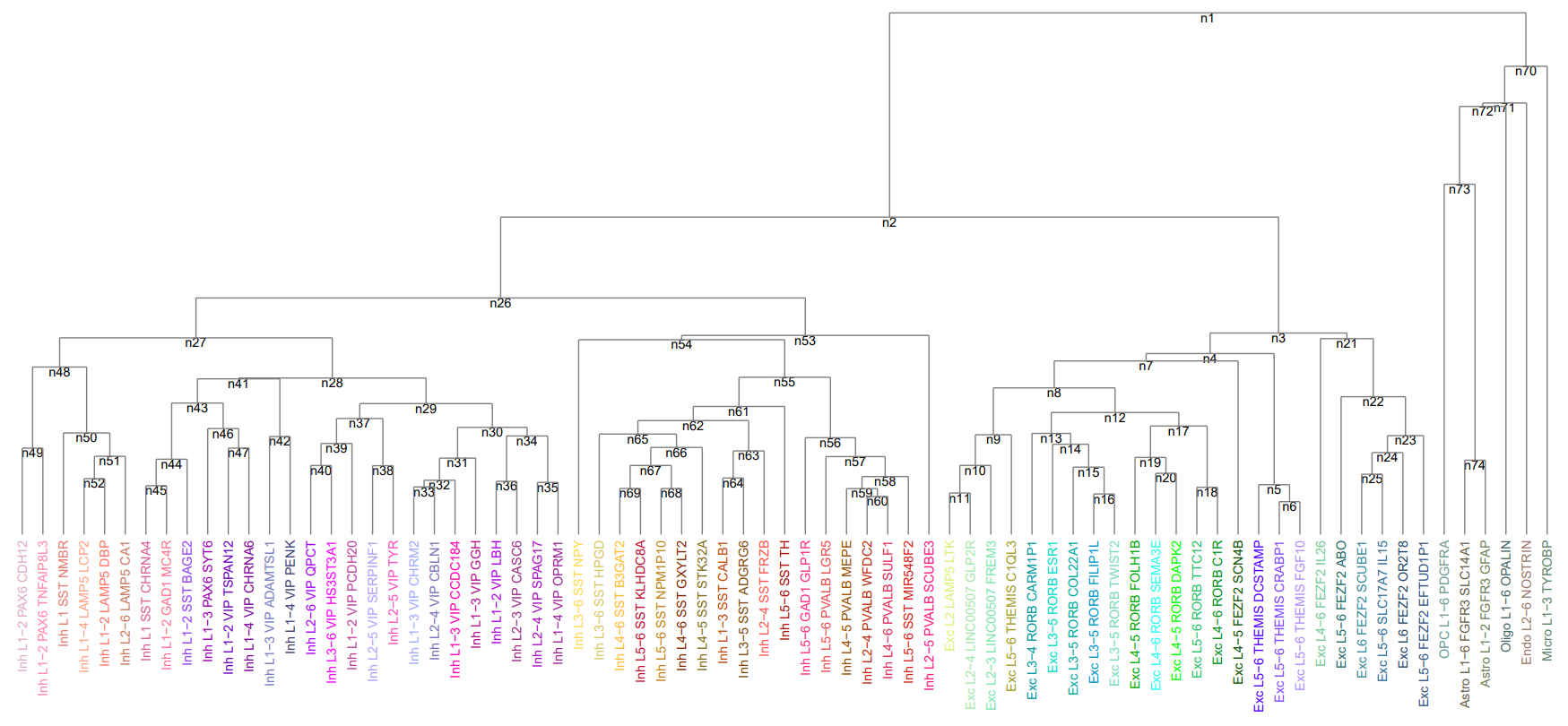
We usually need to select a branch for analysis, which helps us investigate on gene differential expressions on cell subtypes. This package sctreeshap constructs a data structure, which helps us quickly filter data whose clusters are under a specific branch (or in a specific cluster set). Moreover, it can run shap automatically to indicate marker genes.
Github repo:
https://github.com/ForwardStar/sctreeshap
Resolve dependency error:
- Replace
pathlibwithpathlib2sincepathlibhas no longer been supported in Python 3.10; - Remove deprecated sklearn dependency.
Bug fixes: fix function geneFiltering error for the deprecated pandas.DataFrame.iteritems() function.
- Custom model supported.
In v0.7.0--v0.7.4, we mainly worked for supporting probability output for shap values. This is not stable and could not deal with a large feature set. Therefore, try using geneFiltering() before setting output as probability.
A sample code is as follows:
from sctreeshap import sctreeshap
sample = sctreeshap()
sample_dataset = sample.loadDefault()
print(sample_dataset)
## Select non-neuron branch
sample_dataset = sample.selectBranch(sample_dataset, 'n70')
sample_dataset = sample.geneFiltering(sample_dataset, 0.3)
## Run explainer
sample.explainBinary(sample_dataset, 'Micro L1-3 TYROBP', shap_params = {
"model_output": 'probability', # set output mode as 'probability'
"bar_plot" : True,
"beeswarm" : True,
"decision_plot" : True
})
sample.explainMulti(sample_dataset, shap_params = {
"model_output" : 'probability', # set output mode as 'probability'
"bar_plot" : True,
"beeswarm" : True,
"decision_plot" : True
})While in v0.7.5, we supported custom models in explainBinary() and explainMulti(). Currently, there is a new parameter model in the two function. You can set model='XGBClassifier', model='RandomForestClassifier' or model='DecisionTreeClassifier'. However, in the most cases the default XGBClassifier has the best accuracy. You can also set the model as one you defined, like some neuron networks, which may even outperform xgboost models. We do not ensure that shap explainer supports your custom model.
Directly install by pip:
pip install sctreeshapOr by conda:
conda create -n sctreeshap python=3.8
conda activate sctreeshap
pip install sctreeshapAn example dataset, human brain MTG cell type, can be analyzed as default:
# Run in Jupyter Notebook
from sctreeshap import sctreeshap
sample = sctreeshap()
sample_dataset = sample.loadDefault()
print(sample_dataset)
## Select non-neuron branch
sample_dataset = sample.selectBranch(sample_dataset, 'n70')
## Run explainer
sample.explainMulti(sample_dataset)The further details in the process are discussed in the following documents.
A sctreeshap object construction needs a python dict reflecting the tree structure. Here is an example of the cluster tree above.
from sctreeshap import sctreeshap
tree_arr = {
"n1": ('n2', 'n70'),
"n2": ('n26', 'n3'),
"n3": ('n4', 'n21'),
"n4": ('n7', 'n5'),
"n5": ('Exc L5-6 THEMIS DCSTAMP', 'n6'),
"n6": ('Exc L5-6 THEMIS CRABP1', 'Exc L5-6 THEMIS FGF10'),
"n7": ('n8', 'Exc L4-5 FEZF2 SCN4B'),
"n8": ('n9', 'n12'),
"n9": ('n10', 'Exc L5-6 THEMIS C1QL3'),
"n10": ('n11', 'Exc L2-3 LINC00507 FREM3'),
"n11": ('Exc L2 LAMP5 LTK', 'Exc L2-4 LINC00507 GLP2R'),
"n12": ('n13', 'n17'),
"n13": ('Exc L3-4 RORB CARM1P1', 'n14'),
"n14": ('Exc L3-5 RORB ESR1', 'n15'),
"n15": ('Exc L3-5 RORB COL22A1', 'n16'),
"n16": ('Exc L3-5 RORB FILIP1L', 'Exc L3-5 RORB TWIST2'),
"n17": ('n19', 'n18'),
"n18": ('Exc L5-6 RORB TTC12', 'Exc L4-6 RORB C1R'),
"n19": ('Exc L4-5 RORB FOLH1B', 'n20'),
"n20": ('Exc L4-6 RORB SEMA3E', 'Exc L4-5 RORB DAPK2'),
"n21": ('Exc L4-6 FEZF2 IL26', 'n22'),
"n22": ('Exc L5-6 FEZF2 ABO', 'n23'),
"n23": ('n24', 'Exc L5-6 FEZF2 EFTUD1P1'),
"n24": ('n25', 'Exc L6 FEZF2 OR2T8'),
"n25": ('Exc L6 FEZF2 SCUBE1', 'Exc L5-6 SLC17A7 IL15'),
"n26": ('n27', 'n53'),
"n27": ('n48', 'n28'),
"n28": ('n41', 'n29'),
"n29": ('n37', 'n30'),
"n30": ('n31', 'n34'),
"n31": ('n32', 'Inh L1-3 VIP GGH'),
"n32": ('n33', 'Inh L1-3 VIP CCDC184'),
"n33": ('Inh L1-3 VIP CHRM2', 'Inh L2-4 VIP CBLN1'),
"n34": ('n36', 'n35'),
"n35": ('Inh L2-4 VIP SPAG17', 'Inh L1-4 VIP OPRM1'),
"n36": ('Inh L1-2 VIP LBH', 'Inh L2-3 VIP CASC6'),
"n37": ('n39', 'n38'),
"n38": ('Inh L2-5 VIP SERPINF1', 'Inh L2-5 VIP TYR'),
"n39": ('n40', 'Inh L1-2 VIP PCDH20'),
"n40": ('Inh L2-6 VIP QPCT', 'Inh L3-6 VIP HS3ST3A1'),
"n41": ('n43', 'n42'),
"n42": ('Inh L1-3 VIP ADAMTSL1', 'Inh L1-4 VIP PENK'),
"n43": ('n44', 'n46'),
"n44": ('n45', 'Inh L1-2 SST BAGE2'),
"n45": ('Inh L1 SST CHRNA4', 'Inh L1−2 GAD1 MC4R'),
"n46": ('Inh L1-3 PAX6 SYT6', 'n47'),
"n47": ('Inh L1-2 VIP TSPAN12', 'Inh L1-4 VIP CHRNA6'),
"n48": ('n49', 'n50'),
"n49": ('Inh L1-2 PAX6 CDH12', 'Inh L1-2 PAX6 TNFAIP8L3'),
"n50": ('Inh L1 SST NMBR', 'n51'),
"n51": ('n52', 'Inh L2-6 LAMP5 CA1'),
"n52": ('Inh L1-4 LAMP5 LCP2', 'Inh L1-2 LAMP5 DBP'),
"n53": ('n54', 'Inh L2-5 PVALB SCUBE3'),
"n54": ('Inh L3-6 SST NPY', 'n55'),
"n55": ('n61', 'n56'),
"n56": ('Inh L5-6 GAD1 GLP1R', 'n57'),
"n57": ('Inh L5-6 PVALB LGR5', 'n58'),
"n58": ('n59', 'Inh L5-6 SST MIR548F2'),
"n59": ('Inh L4-5 PVALB MEPE', 'n60'),
"n60": ('Inh L2-4 PVALB WFDC2', 'Inh L4-6 PVALB SULF1'),
"n61": ('n62', 'Inh L5-6 SST TH'),
"n62": ('n65', 'n63'),
"n63": ('n64', 'Inh L2-4 SST FRZB'),
"n64": ('Inh L1-3 SST CALB1', 'Inh L3-5 SST ADGRG6'),
"n65": ('Inh L3-6 SST HPGD', 'n66'),
"n66": ('n67', 'Inh L4-5 SST STK32A'),
"n67": ('n69', 'n68'),
"n68": ('Inh L5-6 SST NPM1P10', 'Inh L4-6 SST GXYLT2'),
"n69": ('Inh L4-6 SST B3GAT2', 'Inh L5-6 SST KLHDC8A'),
"n70": ('n71', 'Micro L1-3 TYROBP'),
"n71": ('n72', 'Endo L2-6 NOSTRIN'),
"n72": ('n73', 'Oligo L1-6 OPALIN'),
"n73": ('OPC L1-6 PDGFRA', 'n74'),
"n74": ('Astro L1-6 FGFR3 SLC14A1', 'Astro L1-2 FGFR3 GFAP')
}
Sample = sctreeshap(tree_arr)The keys of the dict represent the name of each tree node, while the values represent the children of each node (the tree can be either binary or multi-children). Note that clusters do not need to be assigned as a key, since they are the leaf nodes in the tree and do not have children.
Then you can read in data by:
data = Sample.readData('samplefile.csv', output='DataFrame')which can recognize .csv, .pkl, .h5ad, .loom, and .xlsx file, and will return an AnnData or DataFrame object.
For the cluster tree in the beginning of this documentation, the corresponding dataset can be loaded by:
data = Sample.loadDefault()After reading in data, you can select a branch in the cluster tree. Cells with clusters not under the branch will be filtered:
# Select non-neuron branch: n70
data = Sample.selectBranch(data, 'n70')You can also filter low-expressed genes, housekeeping genes and general genes by:
prefix = ["MT", "RPS", "RPL", "HSP", "HLA"]
housekeeping = Sample.loadHousekeeping('human')
data = Sample.geneFiltering(data, min_partial=0.3, gene_set=housekeeping, gene_prefix=prefix, use_PCA=False)
print(data)Then genes expressed in <30% cells will be filtered. Genes in gene_set or with prefix in gene_prefix will also be filtered.
You can merge clusters under a branch if needed:
data = Sample.mergeBranch(data, 'n73')This relabels cells with cluster ['OPC L1-6 PDGFRA', 'Astro L1-6 FGFR3 SLC14A1', 'Astro L1-2 FGFR3 GFAP'] as n73.
Note: this part is recommended to run in jupyter notebook.
After reading in the data and filtering, you can build multi-classification model and generate shap figures:
Sample.explainMulti(
data,
use_SMOTE=False,
nthread=48, # multi-thread
shap_params={
"max_display": 10,
"bar_plot": True,
"beeswarm": False,
"decision_plot": False
}
)or build binary-classification model and generate shap figures:
Sample.explainBinary(
data,
cluster_name='Micro L1-3 TYROBP',
use_SMOTE=False,
nthread=48, # multi-thread
shap_params={
"max_display": 10,
"bar_plot": True,
"beeswarm": True,
"force_plot": False,
"heat_map": False,
"decision_plot": False
}
)After running explainBinary() or explainMulti(), you can run:
shap_values = Sample.getShapValues()
marker_genes = Sample.getTopGenes()to get shapley values and marker genes (with top absolute mean shap values).
For more functions, you can refer to the documentations by printing the help function out:
print(Sample.help('documentations'))and query the details of a function by:
function_name = 'readData' # Can be whatever the function in the class
print(Sample.help(function_name))