
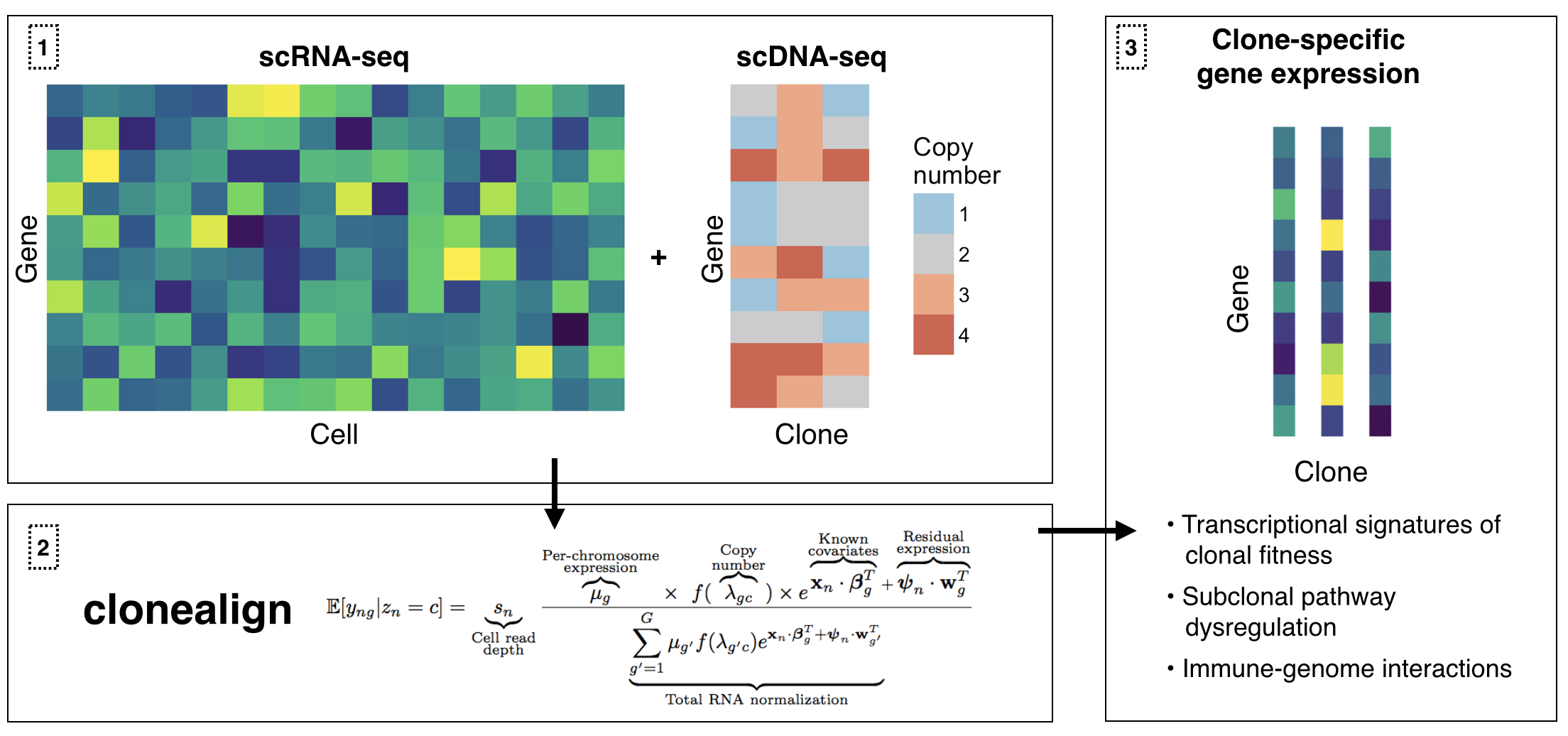
clonealign assigns single-cell RNA-seq expression to cancer clones by probabilistically mapping RNA-seq to clone-specific copy number profiles using reparametrization gradient variational inference. This is particularly useful when clones have been inferred using ultra-shallow single-cell DNA-seq meaning SNV analysis is not possible.
Clonealign version 2.0 comes with several updated modelling features. In particular:
- A multinomial likelihood that vastly increases runtime and removes the need for custom size factors
- Multiple restarts through the
run_clonealignfunction, where the final fit is chosen as that which maximizes the ELBO
For more info see the NEWS.md file.
- Introduction to clonealign Overview of
clonealignincluding data preparation, model fitting, plotting results, and advanced inference control - Preparing copy number data for input to clonealign Instructions for taking region/range specific copy number profiles and converting them to gene and clone specific copy numbers for input to clonealign
clonealign is built using Google's Tensorflow so requires installation of the R package tensorflow. The versioning of Tensorflow and Tensorflow probability currently breaks the standard installation, so the following steps must be taken:
install.packages("tensorflow")
tensorflow::install_tensorflow(extra_packages ="tensorflow-probability", version="2.1.0")
install.packages("devtools") # If not already installed
install_github("kieranrcampbell/clonealign")clonealign accepts either a cell-by-gene matrix of raw counts or a SingleCellExperiment with a counts assay as gene expression input. It also requires a gene-by-clone matrix or data.frame corresponding to the copy number of each gene in each clone. The cells are then assigned to their clones by calling
cal <- clonealign(gene_expression_data, # matrix or SingleCellExperiment
copy_number_data) # matrix or data.frame
print(cal)A clonealign_fit for 200 cells, 100 genes, and 3 clones
To access clone assignments, call x$clone
To access ML parameter estimates, call x$ml_params
print(head(cal$clone))[1] "B" "C" "C" "B" "C" "B"
Kieran R Campbell, University of British Columbia