I am a visual thinker, and knowing only the basics of what I could gather on the details of pair-end short read sequencing in a week, I found a really nice paper and python package for paired-end short read analysis and QC. From what I was able to gather, QC can be controlled for based on a few criteria, of which I chose three which most differentiated the two processed files presented in the assigned task. Based on the plots presented below, which use the three criteria to compare between the processing methods, I determined CH to be the superior method for such analysis, for its nice content distribution, CG count distribution and most tellingly I think, the correlation in both reads between forward and reverse strand bias. From the perspective of a data scientist as well as a biologist, I should think the high correlation between forward and reverse strands a hallmark of accurate sequencing.
Shifu Chen, Tanxiao Huang, Yanqing Zhou, Yue Han, Mingyan Xu and Jia Gu. AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinformatics 2017 18(Suppl 3):80 https://doi.org/10.1186/s12859-017-1469-3
| read | AH method | CH method |
|---|---|---|
| 1 | 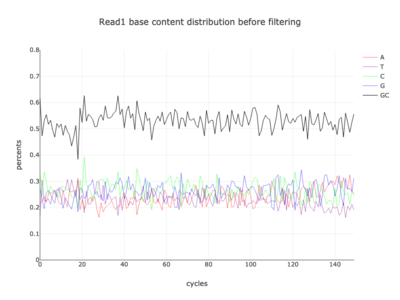 |
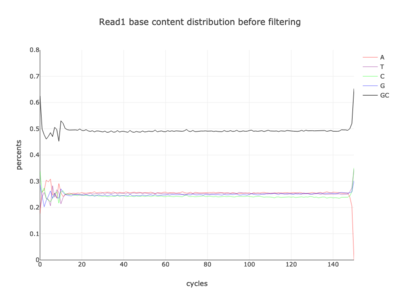 |
| 2 | 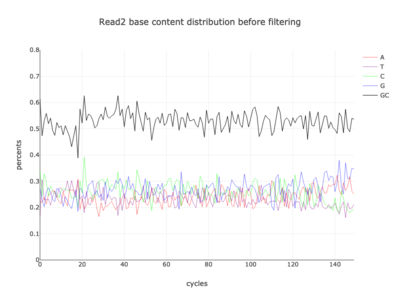 |
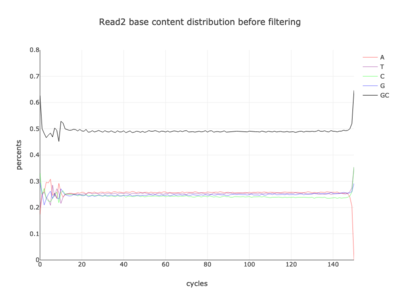 |
| read | AH method | CH method |
|---|---|---|
| 1 | 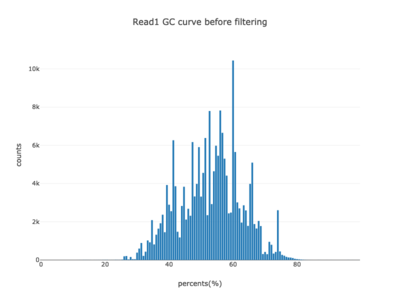 |
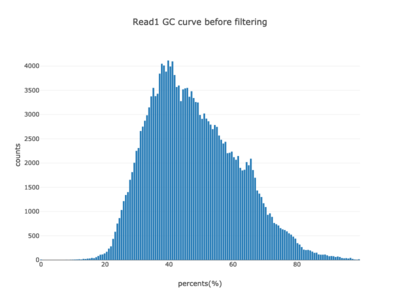 |
| 2 | 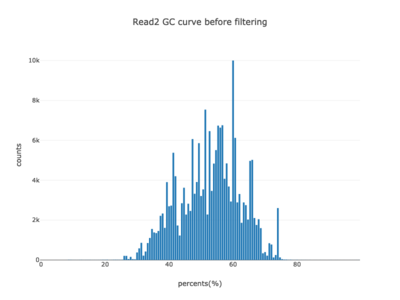 |
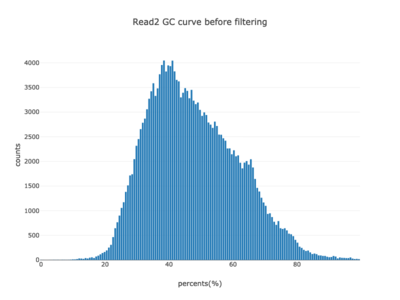 |
| read | AH method | CH method |
|---|---|---|
| 1 | 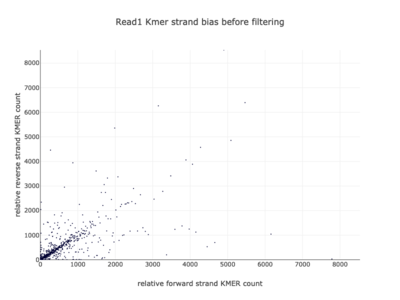 |
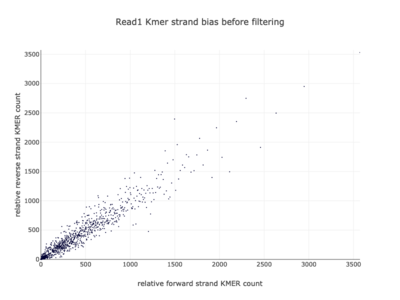 |
| 2 | 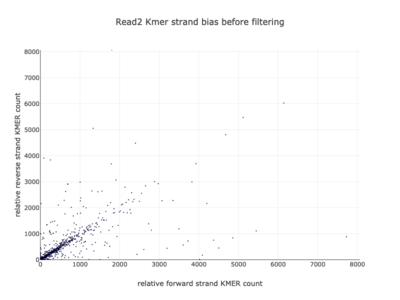 |
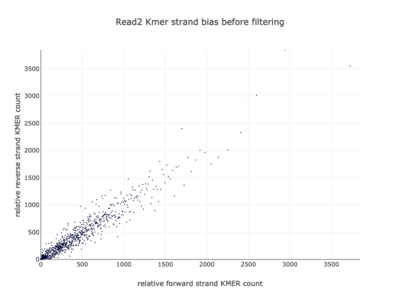 |