Add transform/SMIRKS support to CDK. #916
Conversation
|
Sounds good, maybe a week or so for comment on the API - until release though it’s still changeable. I will fix the code smells and try and get some more test coverage |
|
@uli-f incase you didn’t get pinged on it |
|
@johnmay Thanks for pinging me on this. Thank you so much for the great work! Looks good to me at first glance 😃 I like the Running a bit behind everything, but will def take a closer look this week 👀 |
Shall I send out a tweet? |
No I will add some more doc first then do the mailing list (and maybe tweet) |
|
Had a closer look. Few comments:
|
|
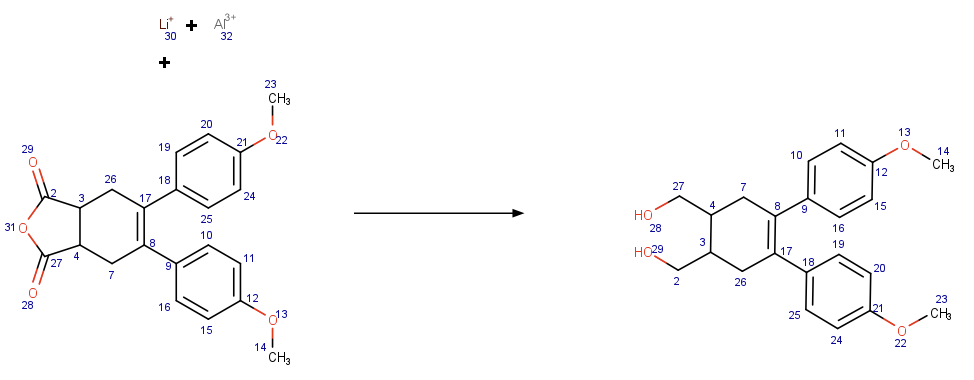
Given the reaction above and the SMILES I added the following method to If I understand correctly the features h, X, and v that are used in this SMIRKS are currently not covered...? |
|
I'll look into it, but doing RDKit UGM talk this week and next (presenting). You can't really have X and v on the right hand side... SMIRKS doesn't work like this :-) The query matches so possibly something else going on. Here is a better way to write the same thing. The error i get is:
Which means atoms :7 and :2 are already bonded - which we can see is the case. This looks to be a bug in the perception of the transform. |
Thanks for pointing that out. I definitely need to take a closer look at SMIRKS and how we use them.
Thanks again. Seeing that already makes me understand a lot better how SMIRKS are supposed to be used. I am happy to put in a few more tests and see how that goes. |
|
Fixed... the bond ordering swapping was not handled correctly. Can you add this as a test case. Something simpler like:
would also have shown the issue. |
Thank you for fixing this 😃 PR #917 Will add more test cases tomorrow. |
|
I discovered that the Enum
Given the transformation patterns I am working with I would definitely appreciate seeing this one coming alive if it adds implicit hydrogens after bond and/or charge changes to an atom. Not quite sure what the expectation would be if the atomic number changes though...? |
|
Doh I didn't mean to check in the options yet as they don't do anything. I really am hesitant about recalculate H, Evan Bolton at pubchem likes the term RoboChemistry :-) |
Okay, totally understand the hesitancy of recalculating the hydrogens. I am wondering if it would be possible to keep track of any changes per atom (bond changes, bond order changes, charge changes) to update the hydrogen count in a more sophisticated way than just recalculating...? Probably a project for another day! |
|
Wait for my ACS talk and I will show the Wild West of SMIRKS bespoke variations. I think having another variant, does not help the situation, however some middle ground might be the ability to take a smirks and improve it by inferring where the hydrogens must be. |
|
Just pushed another PR #918 with a few more test cases. |
Agreed. I am probably just dreaming the dream of meaningful/sensible RoboChemistry :) |
|
Are you manually writing these SMIRKS or generating them, if the later then can make the generate write them better. |
The truth is somewhere in the middle here: The rules are generated automatically, but I edit the RHS to make them valid smirks as you pointed out above that there is no x and v on the RHS in smirks, but only changes to # hydrogens, isotope, charge, atom label (and bond order) for a particular atom. My intention was to add more complex examples instead of testing individual properties / transformation ops. I am very happy to come up with more relevant / correctly written examples if you point me in the right direction. |
|
Had a closer look at the smirks. Do you want me to
Irrespective of the above: I would consider it worthwhile having quirky but valid expressions in the test set. |
|
I played a bit with the different With
But maybe this is more about my expectation being off 🤷🏼 |
|
Matches https://www.daylight.com/dayhtml/javadocs/index.html?com/daylight/Transform.html https://www.daylight.com/dayhtml/doc/man/man3/dt_xtransform.html xtransform is the only one you apply them all at once, the others have overlaps so you need to rematch. |
Sorry about that. It has been a while since I've last used smirks. I will do some reading first so that I can chip in with more reasonable/meaningful contributions here. |
|
What would expect the unique/all options. As described in the doc these are the "iterate possible matches options" and control which matches you apply them to. You can only do a single pass since otherwise (and on non overlapping matches) you could get into an infinite loop of changing things. Consider We could constantly apply this and never end. Such a functionality is possibly useful the the nice thing is you can always build that on top of the "match all" and "match unique" primitives. :-) |
|
Mostly finished my RDKit talk prep now so will try and catch up on this PR/others |
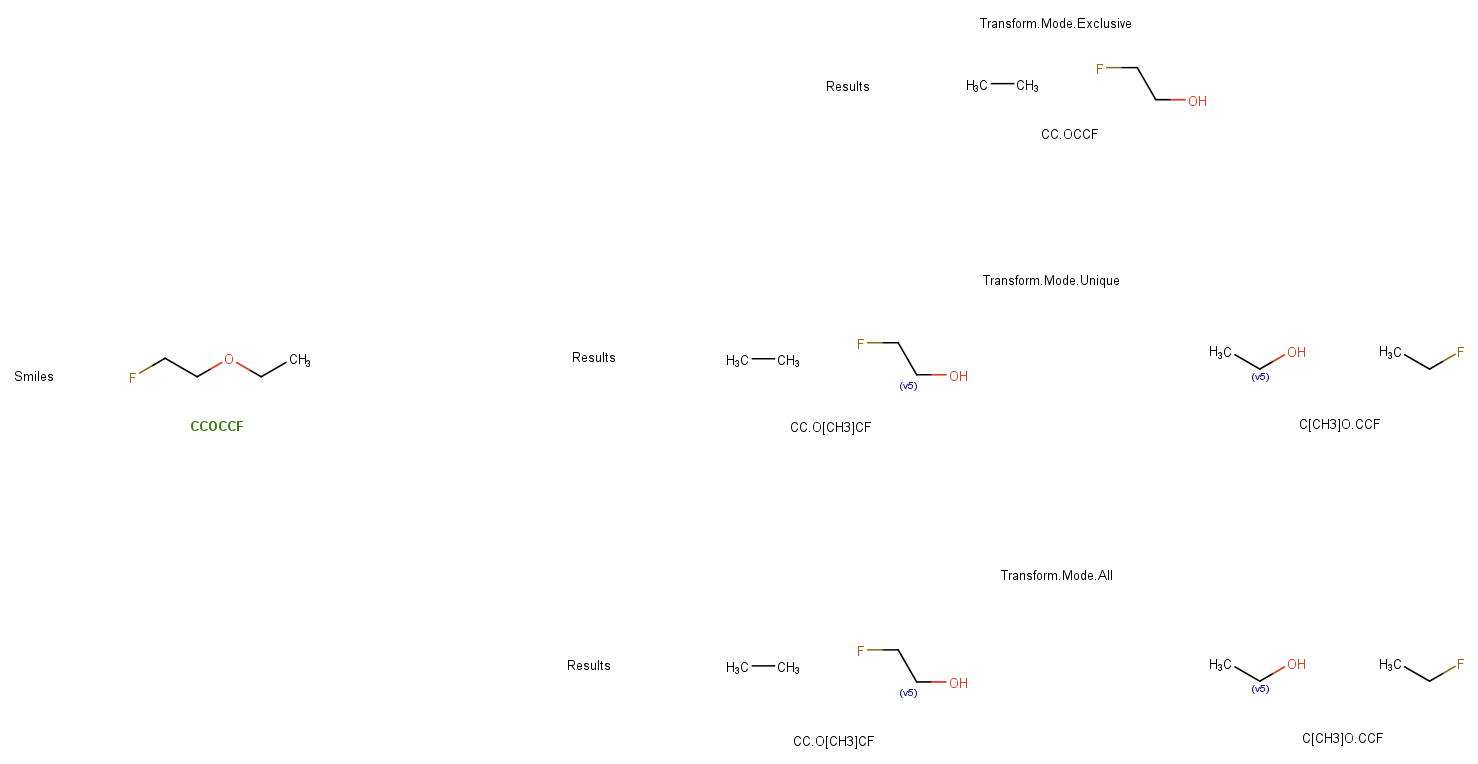
Thank you, I understand this part of the matching and transforming and the associated issue of infinite loops. I am sorry that I did not make it clear enough what surprised me about the result. SMILES: CCOCCF SMILES: CCOCCF Now, if I run the following smirks / smiles combo I get the result I expect: SMILES: CCOCCF Is it correct that the result of the last example could be either CC.OCCF or CCO.CCF depending on how CDK perceives the order of the atoms in the smiles? I expected to remove the bond between O:1 and C:2 using the dot, but that is not what is happening when using Transform.Mode.Unique and Transform.Mode.All. So I end up with transform results that have invalid valences as the CH3 ends up not being terminal. However, if works as expected with Transform.Mode.Exclusive. So it seems like I still don't quite understand something fundamental here.
Okay, I think I understand now what you mean here. I used the Daylight Toolkit years ago and I think in my mind I was looking for that limit argument they have which allows you to limit the number of times the transform is applied with For my application getting the transforms of non-overlapping matches individually might be very helpful. A bit similar to this depiction by Daylight at https://daylight.com/daycgi_tutorials/smirks_examples.html:
|
|
I tried another smirks that carries out a nitro group conversion: With both Is that expected because it makes sense given how the transforms are carried out? So it would be up to the caller of the transform method to check for identical transform results? |
|
I fixed the lazy transform a while ago: Timings are now: I also confirmed this one is OK: |
|



|
I looked more at the javadoc documentation and the API than what happens in the methods. API lgtm, documentation is very helpful. Is there a good place to put the excellent overview of your original comment so that it's not lost once this PR is closed? Would it make sense to put it somewhere in the javadoc? |
|
Oh, if only we still had For eternity we have http://web.archive.org/web/20240306143834/https://github.com/cdk/cdk/pull/916#issue-1385176707 (well, at least until Nov 6) |
|
@uli-f Possibly JavaDoc but also we can add a wiki page (https://github.com/cdk/cdk/wiki) - Egon also has/had the CDK book which has some nice example. Something Roger has emphasised to me over the years is API documentation is separate to the user manual (how to use it). Much like the CDK book and the RDKit book. @egonw what happens on Nov 6? |
It's the "day after". I will try to include the examples in the book, good idea! |
@johnmay Never really thought about this too much, sounds like a good idea.
@egonw Thanks egon, let me know if you need any help with this, I'd be happy to contribute. |
This patch provides the generic/low-level data structures and representations to describe a molecular transform. A conveient way of describing transforms is the SMIRKS syntax which is supported via functionality in the 'cdk-smarts' module.
Transforms are useful for standardisation, library generation, and retro synthesis.
Quick start
cdk-smartsmoduleTransformorSmirksTransformdepending on if you want it to automatically ensure correct aromaticity/ring flags.Features
The atomic number, hydrogen count, charge, and mass can be modified on mapped atoms
There is inconsistency in other implementations about how/when these properties get removed. For example
[OH1:1]>>[O:1]may remove the hydrogen count. CDK's implementation follows a simpler system where by you must explicitly specify you want something removed (zero'd).If the right-hand side has an expression it is examined to see if the property being modified is consistent. The safer portable SMIRKS it is recommend you stick to SMILES
[<mass>?<symbol><hcnt>?<chg>?:<map>]on the right-hand side.Hydrogens are not automatically adjusted, so if you change a property like charge or add/remove bonds you should adjust the hydrogen count accordingly. Subset atoms (B, C, N, O, P, S, F, Cl, Br, I) that are added (unmapped on right-hand side) not in square brackets will automatically set the implicit hydrogen count.
Although it is possible to explicitly set the number of hydrogens with
H<cnt>it is more portable/powerful to use explicit hydrogens in the SMIRKS to adjust the hydrogen count.Note SMIRKS transform does not need the hydrogens to be explicit in the molecule being modified, the transform is compiled and works on either hydrogen counts or explicit hydrogens.
Stereochemistry on atoms with a single point change will persist their stereochemistry.
Multiple changes will result in the stereochemistry being lost
API Overview
The low-level API in the 'cdk-isomorphism' module works with a substructure pattern and ops which change the atoms matched by the pattern. We therefore run a transform by matching the pattern to the molecule, then running the ops over the atoms matched by the pattern. The atoms in the molecule being transformed are permuted/ordering to be the same as the query (actually idx+1) such that the op-code parameters are consistent.
Future additions
Options
Testing other implementations there are some options that may be useful and flags on how things could be interpreted. I've put in the most sensible defaults in (IMO) and the flags would therefore change the behaviour away from that. A nice feature is we can then say "parse this as RDKIT compatibility mode". Although I am planning an ACS talk next spring which will highlight some of the existing behaviours of other implementations are inconsistent/ambiguous.
I've mainly not decided whether to do this via integer bitmaps or enums or another mechanism. Here is the enum option I was toying with: