⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⢀⣀⣤⣤⣴⣶⡆⠀⣶⣶⣦⣤⣤⣄⠀⢠⣾⣿⣦
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⣀⡀⢰⣿⣿⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⣿⡄⠘⢿⣿⠟
⠀⠀⠀⠀⠀⠀⠀⠀⢀⣴⣾⣿⣧⠀⢻⣿⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⣿⣿⠂⢠⣠⣤⣶⣦⡀
⠀⠀⠀⠀⠀⠀⣠⣾⣿⣿⣿⣿⣿⣇⠈⢿⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⣿⠏⢀⣿⣿⣿⣿⣿⣿⣦⡀
⠀⠀⠀⠀⠀⠘⢿⣿⣿⣿⣿⣿⣿⣿⡆⠘⣿⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⣿⡏⠀⣾⣿⣿⣿⣿⣿⣿⡿⠋⢀
⠀⠀⠀⣴⣿⣦⡀⠙⢿⣿⣿⣿⣿⣿⣿⡄⠸⣿⣿⣿⣿⡇⠀⣿⣿⣿⣿⡟⠀⣼⣿⣿⣿⣿⣿⣿⠟⢀⣴⣿⣧
⠀⠀⣼⣿⣿⣿⣿⣦⡀⠙⢿⣿⣿⣿⣿⣷⡀⢹⣿⣿⣿⡇⠀⣿⣿⣿⡿⠁⣰⣿⣿⣿⣿⣿⠟⢁⣴⣿⣿⣿⣿⣧
⠀⣼⣿⣿⣿⣿⣿⣿⣿⣦⡀⠙⢿⣿⣿⣿⣧⠀⢻⣿⣿⡇⠀⣿⣿⣿⠃⢰⣿⣿⣿⣿⠟⢁⣴⣿⣿⣿⣿⣿⣿⣿⣧
⠀⠛⠿⢿⣿⣿⣿⣿⣿⣿⣿⣦⡀⠙⢿⣿⣿⣇⠀⢿⣿⡇⠀⣿⣿⠇⢠⣿⣿⣿⠟⢁⣴⣿⣿⣿⣿⣿⣿⣿⣿⡿⠟⠃
⣼⣶⣤⣄⡈⠙⠛⠿⣿⣿⣿⣿⣿⣦⡀⠙⢿⣿⡆⠈⠛⠃⠀⠛⠋⠀⣾⣿⠟⠁⣠⣾⣿⣿⣿⣿⡿⠿⠛⠉⣀⣠⣴⣶⡄
⣿⣿⣿⣿⣿⣿⣶⣤⣀⡉⠙⠻⢿⣿⣿⣦⠀⠁⠀⠀⠀⠀⠀⠀⠀⠀⠈⠁⢠⣾⣿⣿⠿⠛⠋⢁⣠⣴⣶⣿⣿⣿⣿⣿⣿⡀
⢻⣿⣿⣿⣿⣿⣿⣿⣿⣿⣷⣦⣤⣀⠉⠃⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠈⠋⢁⣀⣤⣶⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣧
⢸⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⡏⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠘⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⡟⠁ ⣠⣶⣄
⠀⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⡄⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⣤⡄⠀⢻⣿⣿⡿
⠀⠘⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⠿⠓⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⣸⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⡿⠂⠀⠙⠋
⠀⠀⠘⣿⣿⣿⣿⣿⠿⠛⠋⢁⣠⣤⣶⣧⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⢰⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⠟⠋
⠀⠀⠀⠘⠟⠋⢉⣀⣤⣶⣿⣿⣿⣿⠟⠁⣠⣦⣄⡀⠀⠀⠀⠀⠀⠀⣤⣦⡈⠻⣿⣿⣿⣿⣿⣿⣿⣿⣿⡿⠛⠁
⠀⠀⠀⠀⠀⠘⢿⣿⣿⣿⣿⣿⠟⠁⣠⣾⣿⣿⣿⣿⣿⣶⣶⣾⣧⠀⢻⣿⣿⣦⡈⠻⣿⣿⣿⣿⡿⠟⠉
⠀⠀⠀⠀⠀⠀⠀⠙⠿⣿⡟⠁⣠⣾⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣇⠈⠿⢿⣿⣿⣦⡈⠻⠛⠉
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠈⠀⠺⢿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⣿⠋⢀⣤⣤⡈⠙⠋⠁
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠉⠙⠛⠻⠿⠿⠿⠿⠿⠿⠇⠀⣿⣿⣿⡇
⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠀⠈⠙⠋
/██ /██ /██ /██ /██████ /██████ /██████ /██ /██ /███████
| ██ | ██ | ███ | ██ |_ ██_/ /██__ ██ /██__ ██ | ███ /███ | ██__ ██
| ██ | ██ | ████| ██ | ██ | ██ \__/ | ██ \ ██ | ████ /████ | ██ \ ██
| ██ | ██ | ██ ██ ██ | ██ | ██ | ████████ | ██ ██/██ ██ | ███████/
| ██ | ██ | ██ ████ | ██ | ██ | ██__ ██ | ██ ███| ██ | ██____/
| ██ | ██ | ██\ ███ | ██ | ██ ██ | ██ | ██ | ██\ █ | ██ | ██
| ██████/ | ██ \ ██ /██████ | ██████/ | ██ | ██ | ██ \/ | ██ | ██
\______/ |__/ \__/ |______/ \______/ |__/ |__/ |__/ |__/ |__/
Authors
Nathan Barros de Souza
Luís Fernando Mercier Franco
NPT/NVT-Monte Carlo Simulation of Mixtures of
Anisomorphic Hard Convex Bodies:
Ellipsoids of revolution, Spherocylinders, and Cylinders
- Disclaimer
- Overview
- Features
- Language
- Building and Compiling
- Data Input
- Files and Folders
- Running the Code
- Reporting Errors
- Citing Us
The authors make no warranties about the use of this software. The authors hold no liabilities for the use of this software. The authors do not recommend the use of this software whatsoever. The algorithm is made freely available to assist researchers in studies involving mixtures of hard convex bodies. All information contained herein regarding any specific methodology does not constitute or imply its endorsement or recommendation by the authors.
This NVT/NPT-Monte Carlo algorithm was designed to evaluate the thermodynamic behavior of mixtures of anisomorphic hard convex bodies (HCB), namely: ellipsoids of revolution (EOR), spherocylinders (SPC), and cylinders (CYL). Here, the HCB interact either through a spherical square well potential (Version 1.3.1) or a purely repulsive hard-core potential, which means that a single check of overlapping configurations is sufficient to validate a random trial move. It is also possible to select a force field to account for the attractive interactions in the perturbed system (NVT only), but they do not affect the probability distribution function of accepting random trial moves. To search for overlapping configurations, different methods were applied depending on the molecular geometry: for mixtures of anisomorphic EOR particles, the Perram-Wertheim (PW) method (J. Comput. Phys., 58, 409-416, 1985) is used; for mixtures of anisomorphic SPC particles, the Vega-Lago (VL) method (Comput. Chem., 18, 55-59, 1994) is used; and for mixtures of CYL particles, a modified version of Lopes et al. (mLCYL) algorithm (J. Chem. Phys., 154, 104902, 2021) is used. We have also implemented a new algorithm to search for overlaps between cylinders and spheres.
This NVT/NPT-Monte Carlo algorithm only treats mixtures of anisomorphic HCB, that is, mixtures of HCB with same geometry but different aspect ratios*. Mixtures of isomorphic HCB are also possible. The geometries are illustrated below.
| Oblate Ellipsoids of Revolution | Spheres (Degenerate) | Prolate Ellipsoids of revolution |
 |
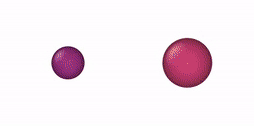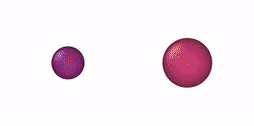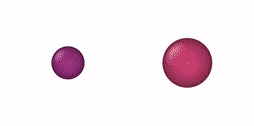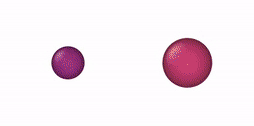 |
 |
| Thin Spherocylinders | Spheres (Degenerate) | Thick Spherocylinders |
 |
|
 |
| Oblate Cylinders | Equilateral Cylinders | Prolate Cylinders |
 |
 |
 |
*The mLCYL algorithm also searches for overlapping configurations between cylinders and spheres (Version 1.1.0).
This NVT/NPT-Monte Carlo algorithm can be used, e. g., for mixtures of oblate and prolate ellipsoids of revolution, oblate/prolate ellipsoids of revolution and spheres (degenerate case), thin and thick spherocylinders, thin/thick spherocylinders and spheres (degenerate case), oblate (disk) and prolate (rod) cylinders, oblate/prolate cylinders and equilateral cylinders, and cylinders and spheres. Some NVT-Monte Carlo simulations of these multicomponent mixtures are illustrated below.
| NVT-Monte Carlo (EOR) | NVT-Monte Carlo (SPC) | NVT-Monte Carlo (CYL) |
 |
 |
 |
The following features are supported in the current version:
-
Initial Configurations
- Simple Cube (for pure components and mixtures)
- Body-Centered Cube (for pure components and mixtures)
- Face-Centered Cube (for pure components and mixtures)
- Random Structure (for pure components and mixtures)
-
Ensembles
- Canonical (NVT)
- Isothermal-Isobaric (NPT)
- Isothermal-Isostress (NσT)
-
Moves
- Translation
- Rotation (Quaternion Algebra)
- Cubic Expansions/Compressions (Isotropic)
- Orthorhombic Expansions/Compressions (Anisotropic)
- Triclinic Deformations
-
Lattice Reduction Algorithms
-
Force Fields
- Spherical Square Well (both perturbed and reference system*)
-
Cell Lists
*Version 1.3.1.
The main program, subroutines, modules, and functions contain some explanatory comments and are mainly written in Fortran 95. The user can look for more information on Fortran language here.
Firstly, you must clone this GitHub repository using the command below:
> git clone https://github.com/LESC-Unicamp/Monte-Carlo-Mixtures-of-Ellipsoids-Spherocylinders-Cylinders
After that, change to the repository directory on your device and open the /src/ folder:
> cd Monte-Carlo-Mixtures-of-Ellipsoids-Spherocylinders-Cylinders/src/
The /src/ folder contains two 'Makefiles' that you can use to compile the code. Each of these 'Makefiles' contains different compilation options. You can use either the 'makefile' to compile the standard version of the code or the 'makefile-debug' to compile the debug version of the code.
To compile it, first remove any object and module files using the clean command:
> make clean
Then, to compile the source code, run one of the commands below, depending on the chosen compilation type:
| Compilation type | Command |
|---|---|
| Debug (without OpenMP) | make -f makefile-debug |
| Debug (with OpenMP) | make -f makefile-debug-openmp |
| Standard (without OpenMP) | make -f makefile |
| Standard (with OpenMP) | make -f makefile-openmp |
If your compilation option includes the OpenMP API (Version 1.4.0), you can also choose the number of threads to be used on your simulation via the following command:
> export OMP_NUM_THREADS=X
where X is the number of threads.
Each of these commands will produce an executable with a specific name depending on the chosen compilation type. All executables can be found in the folder /bin/ in the repository directory. Before running the program, there are a few initialization variables and options you need to set up.
Apart from the executable file, the /bin/ folder contains some initialization files that need to be set up.
The Configuration File
ini_config.ini
This file is used to set up the molecular geometry and the molecular configuration, including aditional information on the random structure (if selected). The table below shows some options that can be used to define the configuration parameters:
| Name ______________________ |
String Name _________________________________ |
Definition _______________________________ |
Options _________________________________________________ |
|---|---|---|---|
| Geometry selection | geometry_selection |
Used to select the molecular geometry |
|
| Configuration selection | molecular_configuration |
Used to select the initial configuration |
|
| Unrotated axis | unrotated_axis |
Used to select the unrotated reference axis (initial configurations only) |
|
| Quaternion angle | quaternion_angle |
Used to select the orientation angle in degrees (initial configurations only) | Any FLOAT number |
| Packing fraction ( RND only) |
packing_fraction_rnd |
Used to define the initial volume of the simulation box (random configuration only) NOTE: Smaller packing fractions (larger box volumes) are recommended |
Any positive, non-zero FLOAT number between 0 and 1 |
| Reduced pressure¹ ( RND only) |
pressure_npt_rnd |
Used to correct the initial packing fraction of the simulation box to the target packing fraction (random configuration only) NOTE: Higher pressures are recommended |
Any positive, non-zero FLOAT number |
| Preset configuration | preset_initial_configuration |
Used to replace the current configuration with a previously generated configuration NOTE: This overrides most of the simulation settings |
|
¹P* = Pσ0³/(kBT), where P is the real pressure, kB is the Boltzmann constant, T is the absolute temperature, and σ0 is a reference diameter, such that σ0 = 1Å.
OBS.: When selecting a preset configuration, the user will be asked to enter a 14-character code at some point. This code is part of the name of a configuration file inside the '/bin/Initial_Configuration/' directory. The filename says something like:
[DATE][HOUR]_initconf_[CONFIGURATION]_[GEOMETRY].xyz
The 14-character code is the [DATE][HOUR] descriptor of the filename. You can open this file using any text editor and edit some simulation settings that you find relevant. Please be reminded that editing anything in this file, except the reduced pressure and absolute temperature, may cause the program to detect overlapping configurations in the initial structure.
The Control File
ini_control.ini
This file is used to set up some control variables, such as data printing, seed type, and potential type. The table below shows some options that can be used to define the control parameters:
| Name ______________________ |
String Name _________________________________ |
Definition _________________________________ |
Options _________________________________________________ |
|---|---|---|---|
| Trajectory printing | print_trajectory |
Used to specify whether the trajectory files will be written out or not |
|
| Seed type | fixed_seed |
Used to control the type of pseudorandom number generator seed |
|
| Random number generator | random_number_generator |
Used to select the pseudorandom number generator |
|
| Backup control | backup_file |
Used to specify whether a backup file will be generated every saving_frequency cycles |
|
| Restore backup | restore_backup |
Used to restore data from previous, unfinished simulations |
|
| Potential type (perturbed system) |
perturbed_potential_type |
Used to control the type of potential applied in the perturbed system |
|
| Potential type (reference system) |
full_potential_type |
Used to control the type of potential applied in the reference system |
|
| Cell lists | cell_lists |
Used to improve the simulation performance by considering neighbour cell lists instead of considering the whole system |
|
| Potential energy printing | potential_production_only |
Used to specify whether the potential energy data will be written out during the whole NVT-simulation or only for production-related cycles |
|
| TPT¹ coefficients printing | coefficients_calculation |
Used to specify whether the first- and second-order coefficients of a high-temperature expansion of the Helmholtz free energy will be calculated or not (NVT-simulation only) |
|
¹TPT stands for thermodynamic perturbation theory.
OBS. I: When selecting to restore data from a backup file, the user will be asked to enter a 14-character code at some point. This code is part of the name of two configuration files inside the '/bin/Backup/' directory. The filenames say something like:
[DATE][HOUR]_simulation.backup
[DATE][HOUR]_variables.backup
OBS. II: cell lists are implemented for both the overlap check and potential computation.
The Monte Carlo File
ini_montecarlo.ini
This file is used to set up the number of simulation cycles, maximum displacements, ensemble type, and lattice reduction algorithm (if anisotropic volume changes are considered). The table below shows some options that can be used to define the simulation parameters:
| Name ______________________ |
String Name _________________________________ |
Definition _______________________________ |
Options _________________________________________________ |
|---|---|---|---|
| Number of cycles | max_cycles |
Used to define the maximum number of simulation cycles NOTE: A cycle is defined as N attempts to displace a random particle in the system or a single attempt to change the volume of the box |
Any positive, non-zero INTEGER number |
| Number of equilibration cycles | equilibration_cycles |
Used to define the number of equilibration cycles within the maximum number of cycles | Any positive, non-zero INTEGER number less than the maximum number of cycles |
| Saving frequency | saving_frequency |
Used to define how often simulation results are written out | Any positive, non-zero INTEGER numberNOTE: 1 is the highest frequency, meaning that the results will be written out every simulation cycle |
| Adjustment frequency (movement) |
adjustment_frequency_movement |
Used to define how often movement displacement adjustments are carried out | Any positive, non-zero INTEGER number |
| Adjustment frequency (volume change) |
adjustment_frequency_volume |
Used to define how often volumetric displacement adjustments are carried out | Any positive, non-zero INTEGER number |
| Adjustment frequency (movement) ( RND only) |
adjustment_frequency_rnd_mov |
Used to define how often movement displacement adjustments are carried out (random configuration only) | Any positive, non-zero INTEGER numberNOTE: 1 is the highest frequency, meaning that the displacements will be adjusted every simulation cycle |
| Adjustment frequency (volume) ( RND only) |
adjustment_frequency_rnd_vol |
Used to define how often volumetric displacement adjustments are carried out (random configuration only) | Any positive, non-zero INTEGER numberNOTE: 1 is the highest frequency, meaning that the displacements will be adjusted every simulation cycle |
| Maximum translational displacement | max_translational_displc |
Used to define the maximum translational displacement [+/-] in Å | Any non-zero FLOAT number |
| Maximum translational displacement ( RND only) |
max_translational_displc_rnd |
Used to define the maximum translational displacement [+/-] in Å (random configuration only) | Any non-zero FLOAT number |
| Maximum rotational displacement | max_rotational_displc |
Used to define the maximum rotational displacement [+/-] in radians | Any non-zero FLOAT number |
| Maximum rotational displacement ( RND only) |
max_rotational_displc_rnd |
Used to define the maximum rotational displacement [+/-] in radians (random configuration only) | Any positive, non-zero FLOAT number |
| Maximum isotropic volumetric displacement | max_volumetric_displc_iso |
Used to define the maximum isotropic volumetric displacement [+/-] in ų | Any non-zero FLOAT number |
| Maximum anisotropic volumetric displacement | max_volumetric_displc_aniso |
Used to define the maximum anisotropic volumetric displacement [+/-] in ų | Any non-zero FLOAT number |
| Maximum isotropic volumetric displacement ( RND only) |
max_volumetric_displc_iso_rnd |
Used to define the maximum isotropic volumetric displacement [+/-] in ų (random configuration only) | Any non-zero FLOAT number |
| Maximum anisotropic volumetric displacement ( RND only) |
max_volumetric_displc_aniso_rnd |
Used to define the maximum volumetric displacement [+/-] in ų (random configuration only) | Any non-zero FLOAT number |
| Minimum volumetric displacement ( RND only) |
min_volumetric_displc_rnd |
Used to define the minimum volumetric displacement [+/-] in ų (random configuration only) | Any non-zero FLOAT number such that its absolute value is less than the maximum isotropic and anisotropic volumetric displacements |
| Maximum box distortion | max_box_distortion |
Used to define the maximum box distortion before carrying out a lattice reduction technique | Any positive FLOAT number greater than 1 |
| Maximum box length distortion | max_box_length_ratio |
Used to define the maximum ratio between the lengths of the edges of the box. | Any positive, non-zero FLOAT number |
| Maximum box angle distortion | max_box_angle_degree |
Used to define the maximum deformation angle (in degrees) between the edges of the box in comparison to a perfect cube (90° + x). | Any positive, non-zero FLOAT number |
| Lattice reduction algorithm | lattice_reduction |
Used to define the algorithm that carries out the lattice reduction technique |
|
| Ensemble type | ensemble |
Used to define the statistical ensemble |
|
OBS. I: The number of production cycles is defined automatically by subtracting the number of equilibration cycles from the total number of cycles.
OBS. II: The box distortion is defined as the product of the total surface area and perimeter of the box divided by its volume. We normalize the box distortion factor by dividing it by 72, which corresponds to minimum box distortion possible (perfect cube). In that case, 1 is a perfect cube and higher values represent triclinic or non-cubic orthorhombic structures.
The Potential File
ini_potential.ini
This file is used to set up variables related to the chosen force field. The table below shows some options that can be used to define the force field parameters:
| Name ________________________ |
String Name _______________________ |
Definition _________________________________ |
Options _______________________________________ |
|---|---|---|---|
| Attrative range points Square-well potential |
lambda_points |
Used to define the number of attractive range points for which the potential will be computed | Any positive, non-zero INTEGER number |
| Attrative range values Square-well potential |
lambda_values |
Used to define the attractive range of the potential | Any positive FLOAT number greater than 1 |
| Reduced temperature¹ | reduced_temperature |
Used to define the reduced temperature of the system | Any positive, non-zero FLOAT number |
| Minimum number of blocks | min_blocks |
Block-average parameter: Used to define the minimum number of blocks to compute the statistical inefficiency of a dataset |
Any positive, non-zero INTEGER number |
| Maximum number of blocks | max_blocks |
Block-average parameter: Used to define the maximum number of blocks to compute the statistical inefficiency of a dataset |
Any positive INTEGER number greater than the minimum number of blocks |
¹T* = (kBT)/ϵ, where T is the absolute temperature in Kelvin, kB is the Boltzmann constant, and ϵ is the well depth of the potential.
OBS. I: If the number of attractive range points is greater than 1, the attractive range values must be entered successively on the same respective line, separated by a single space, as shown in the provided *.ini file.
OBS. II: The block-average parameters are used by a block-average subroutine that computes the perturbation coefficients from the potential dataset.
OBS. III: If the square well potential is chosen for the reference system, then only one potential range will be allowed.
The Probabilities File
ini_probabilities.ini
This file is used to set up the probability of trial moves. The table below shows some options that can be used to define the probability parameters:
| Name ______________________ |
String Name _________________________________ |
Definition _______________________________ |
Options _________________________________________________ |
|---|---|---|---|
| Probability of movement | prob_movement |
Used to define the probability a movement (translation/rotation) will occur during a trial move | Any positive FLOAT number between 0 and 1 |
| Probability of movement ( RND only) |
prob_movement_rnd |
Used to define the probability a movement (translation/rotation) will occur during a trial move (random configuration only) | Any positive FLOAT number between 0 and 1 |
| Probability of translation | prob_translation |
Used to define the probability a translation will occur during a trial movement | Any positive FLOAT number between 0 and 1 |
| Probability of translation ( RND only) |
prob_translation_rnd |
Used to define the probability a translation will occur during a trial movement (random configuration only) | Any positive FLOAT number between 0 and 1 |
| Probability of isotropic volume change | prob_volume_change_isotropic |
Used to define the probability an isotropic change will occur during a trial volume change | Any positive FLOAT number between 0 and 1 |
Probability of isotropic volume change (RND only) |
prob_volume_change_isotropic_rnd |
Used to define the probability an isotropic change will occur during a trial volume change (random configuration only) | Any positive FLOAT number between 0 and 1 |
OBS. I: The probability of a volume change is defined automatically by subtracting the probability of movement from 1. Similarly, the probability of rotation is defined by subtracting the probability of translation from 1. Finally, the probability of an anisotropic volume change is defined by subtracting the probability of the isotropic change from 1.
OBS. II: If the NVT ensemble is selected, the probability of movement is replaced by 1. If the NPT ensemble is selected, the probability of movement cannot be 1, which means that the probability of volume change cannot be set to 0.
The Acceptance Ratios File
ini_ratio.ini
This file is used to set up the acceptance ratio thresholds of trial moves. The table below shows some options that can be used to define the threshold parameters:
| Name ______________________ |
String Name _________________________________ |
Definition _______________________________ |
Options _________________________________________________ |
|---|---|---|---|
| Translational threshold | ratio_translation |
Used to define the acceptance threshold of translational moves | Any positive FLOAT number between 0 and 1 |
| Rotational threshold | ratio_rotation |
Used to define the acceptance threshold of rotational moves | Any positive FLOAT number between 0 and 1 |
| Isovolumetric threshold | ratio_volume_iso |
Used to define the acceptance threshold of isotropic volumetric moves | Any positive FLOAT number between 0 and 1 |
| Anisovolumetric threshold | ratio_volume_aniso |
Used to define the acceptance threshold of anisotropic volumetric moves | Any positive FLOAT number between 0 and 1 |
OBS.: Adjustments to maximum diplacements are only made during the equilibration phase for every adjustment_frequency cycles. Let's call the number of accepted moves n_accepted and the number of trialed moves n_trialed. If n_accepted / n_trialed > threshold, the corresponding maximum displacement is increased by 5% of its current value; otherwise, it is decreased by 5% of its current value.
The System File
ini_system.ini
This file is used to set up system-related variables, including geometric properties. The table below shows some options that can be used to define the system parameters:
| Name ______________________ |
String Name _________________________________ |
Definition _______________________________ |
Options _________________________________________________ |
|---|---|---|---|
| Packing fraction | packing_fraction |
Used to define the target packing fraction of an NVT-simulation or the initial packing fraction (initial volume) of an NPT-simulation | Any positive, non-zero FLOAT number between 0 and 1 |
| Number of components | components |
Used to define the number of components in the mixture | Any positive, non-zero INTEGER number |
| Spherical components | spherical_components |
Used to identify the spherical components in the mixture |
|
| Molecular diameter | diameter |
Used to define the molecular diameter in Å of each component in the mixture | Any positive, non-zero FLOAT number |
| Molecular length | length |
Used to define the molecular length in Å of each component in the mixture | Any positive, non-zero FLOAT number |
| Mole fraction | molar_fraction |
Used to define the mole fraction of each component in the mixture | Any positive, non-zero FLOAT number between 0 and 1 |
| Number of particles | number_of_particles |
Used to define the total number of particles | Any positive, non-zero INTEGER number |
| Temperature | absolute_temperature |
Used to define the temperature of the system in K | Any positive, non-zero FLOAT number |
| Reduced pressure¹ | reduced_pressure |
Used to define the reduced pressure (only used in an NPT-simulation) | Any positive, non-zero FLOAT number |
¹P* = Pσ0³/(kBT), where P is the real pressure, kB is the Boltzmann constant, T is the absolute temperature, and σ0 is a reference diameter, such that σ0 = 1Å.
OBS. I: If the number of components is greater than 1, the identification of spherical components, the molecular diameters, molecular lengths, and mole fractions must be entered successively on the same respective line, separated by a single space, as shown in the provided *.ini file.
OBS. II: If, for any reason, the total mole fraction exceeds 1, it will be automatically normalized.
OBS. III: The total number of particles may be slightly adjusted depending on the selected mole fractions.
OBS. IV: The absolute temperature is only required to calculate the real pressure in MPa.
The program organizes the output files and directories automatically. It creates 9 directories in total. To better organize the output files, all directories (or subdirectories) contains a date subfolder, corresponding to the starting date/hour of the simulation.
The Backup/ directory stores the backup files of the current simulation. These files hold information on system variables (number of components, number of particles, etc.) and on simulation variables (particles' positions and orientations, box properties, cell list properties, etc.). The backup files are written out every saving_frequency cycles.
The Box/ directory stores information on the box volume and box length, including the current distortion of the simulation box (only valid for NPT-simulations). This property is written out every saving_frequency cycles.
The Initial_Configuration/ directory stores the preset initial configuration file. It also contains the OVITO/ subdirectory, which holds preformatted information on the position and orientation of particles, as well as their molecular geometry, that can be visualized directly on OVITO software. The OVITO/ subdirectory also contains an 'RND' configuration file that is constantly updated to allow you to keep up with the development of the random configuration.
The Order_Parameter/ directory stores the nematic order parameter. This property is written out every saving_frequency cycles.
The Perturbed_Coefficient/ directory stores the calculated TPT coefficients and full Helmholtz free energy of the perturbed system. These properties are written out at the end of the simulation. If the square-well potential is selected, there will also be a subfolder for each attractive range point.
The Potential/ directory stores information on the potential energy calculated through the selected force field. This property is written out every saving_frequency cycles. If the control variable potential_production_only is set to Y, then only production cycles will be accounted for. If the square-well potential is selected, there will also be a subfolder for each attractive range point.
The Ratio/ directory stores information on equilibration cycles (acceptance ratio, maximum displacements, etc.). This information is sorted out into three subfolders: the Rotation/ subfolder holds information on rotational moves; the Translation/ subfolder holds information on translational moves; and the Volume/ subfolder holds information on volumetric moves (only valid for NPT-simulations). These properties are only written out every adjustment_frequency equilibration cycles.
The Results/ directory stores relavant results of the NPT-simulation, including the packing fraction, number density, and box volume. These properties are written out every saving_frequency cycles.
The Trajectories/ directory stores the trajectory file containing preformatted information on the position and orientation of particles, as well as their molecular geometry, that can be visualized directly on OVITO software. These properties are written out every saving_frequency cycles.
NOTE: the trajectory is only written out if the parameter print_trajectory is set to Y.
The aforementioned folders are created by executing a shell command via an intrinsic function called SYSTEM which works in Linux operating systems and Linux subsystems. Please note that which shell is used to invoke the command line is system-dependent and environment-dependent. Check this link for more information.
Filenames are based on the information they hold, followed by the packing fraction (NVT-simulations) or reduced pressure (NPT-simulations) and the number of components that were used to create them. To prevent identically named files from being overwritten, a time prefix indicating when the simulation started is added to the file.
Finally, it's time to run the code! After compilation, the executable(s) can be found in the /bin/ folder:
Standard compilation (without OpenMP API)
./mc_mixtures_standard.out
Standard compilation (with OpenMP API)
./mc_mixtures_standard_openmp.out
or
Debug compilation (without OpenMP API)
./mc_mixtures_debug.out
Debug compilation (with OpenMP API)
./mc_mixtures_debug_openmp.out
First, when running the code, a summary of every property previously defined in the *.ini files will be printed out on the screen. If everything is OK, enter "Y" to continue. Otherwise, enter "N" to exit and edit something.
The first thing the algorithm does is create the initial configuration folder and the initial configuration file. If you selected the random structure as the initial configuration, you can also check what stage the code is in while it builds the random configuration. After that, the algorithm creates the remaining folders and subfolders.
Next, the algorithm checks if the initial configuration has any overlapping configurations. This is more relevant when using a preset initial configuration or selecting an SC, BCC, or FCC structure as the initial configuration. If the algorithm detects an overlapping configuration, it'll inform which particles are overlapping before exiting the program.
Next, the property files are created and the real simulation starts. You can see what stage the simulation is at through the progress bar. When the simulation finishes, the program will write out a simulation log. This is automatically done after every simulation. The first simulation creates the simulation log file. Future simulations will only append information to the already existing log file.
If you spot an error in the program files and all other documentation, please submit an issue report using the Issues tab.
N. B. de Souza, J. T. Lopes, L. F. M. Franco. Fluid Ph. Equilibria 2022, 561, 113543. DOI: 10.1016/j.fluid.2022.113543
“With thermodynamics, one can calculate almost everything crudely; with kinetic theory, one can calculate fewer things, but more accurately; and with statistical mechanics, one can calculate almost nothing exactly.” -- Eugene P. Wigner