| date | comments |
|---|---|
2023-12-31 |
true |
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
本周话题:哪些技能/知识使生物信息学不可替代?
现在很多从事湿实验室工作的人开始学习生物信息学来分析他们的数据。那么,在你看来,作为一名生物信息学分析师,要想在就业市场上保持竞争力,应该提升哪些技能或关注哪些领域呢?我们是不是应该更多地关注计算机科学而不是生物学方面的知识。
- 来个大橙子:找好你的定位并重视以及十分重视生物知识的学习。以个人的愚见,我将生物信息工作者分为两大类,一类是bioinformatician/bioinformatics engineer, 着重生物信息技术面,例如开发算法,构建pipeline,建数据库之类。他们往往有比较强的computer science 和coding能力。第二类是bioinformatics scientist, 更看重利用生物信息学手段解决生物学问题。当然我属于后者。我想大部分生物信息工作者也属于这类。对于定位第一类的,我经验不多,无法给出好的建议。对于第二类,我想特别强调生物知识的重要,尤其是当你不想成为工具人。一般来说,在药企里当你对生物学了解更多,就可以和bench scientist 更好的交流,你也更能提供数据分析的独特角度,发现数据中体现的生物学故事。如果你想往上成为管理岗位,更是要如此。这点对大部分在科研院所从事生物学研究的实验室也是如此。我曾经问我们CSO 如何成为一个好的bioinformatics scientist 他的回答就是重视生物学知识的学习,而很多人却过于重视编程或者数据分析技术的学习。我想以后类似于chatGPT等工具的应用更需要你将重心转移到生物的学习上了。
1、Nature | 综合多组学图谱揭示癌症转变过程的表观遗传调控
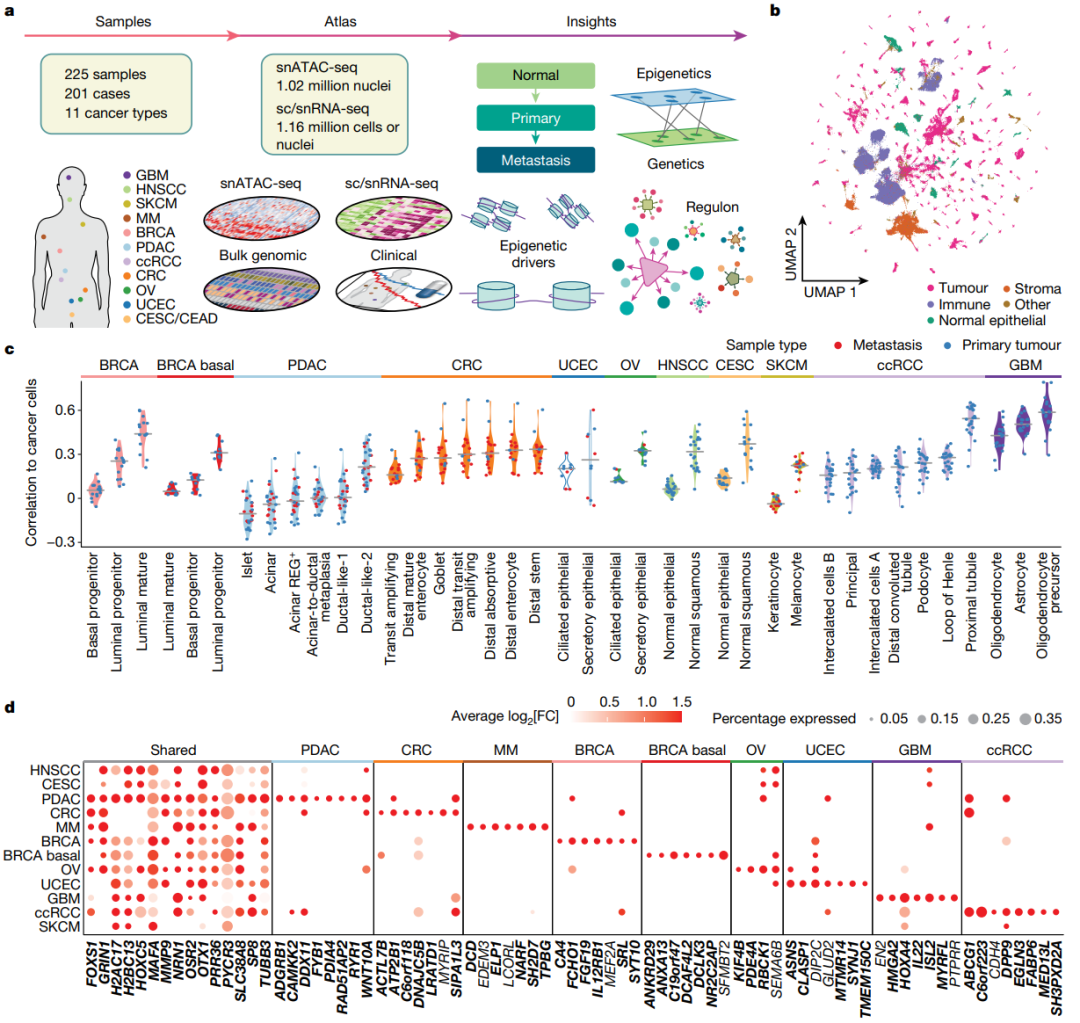
作为NCI人类肿瘤图谱网络(HTAN)的一部分,研究团队从201名患者中收集了225个样本,包括158个原发性肿瘤样本、52个转移性肿瘤样本和15个正常邻近组织(NAT),共涵盖11种癌症类型。研究团队构建了一个综合多组学图确定了与癌症相关的表观遗传驱动因素,并强调了转录因子(TF)作为预后标志物的潜力。
2、Briefings in Bioinformatics | 一种无需参考基因组在转录组数据中识别可变剪切的新方法“MkcDBGAS”
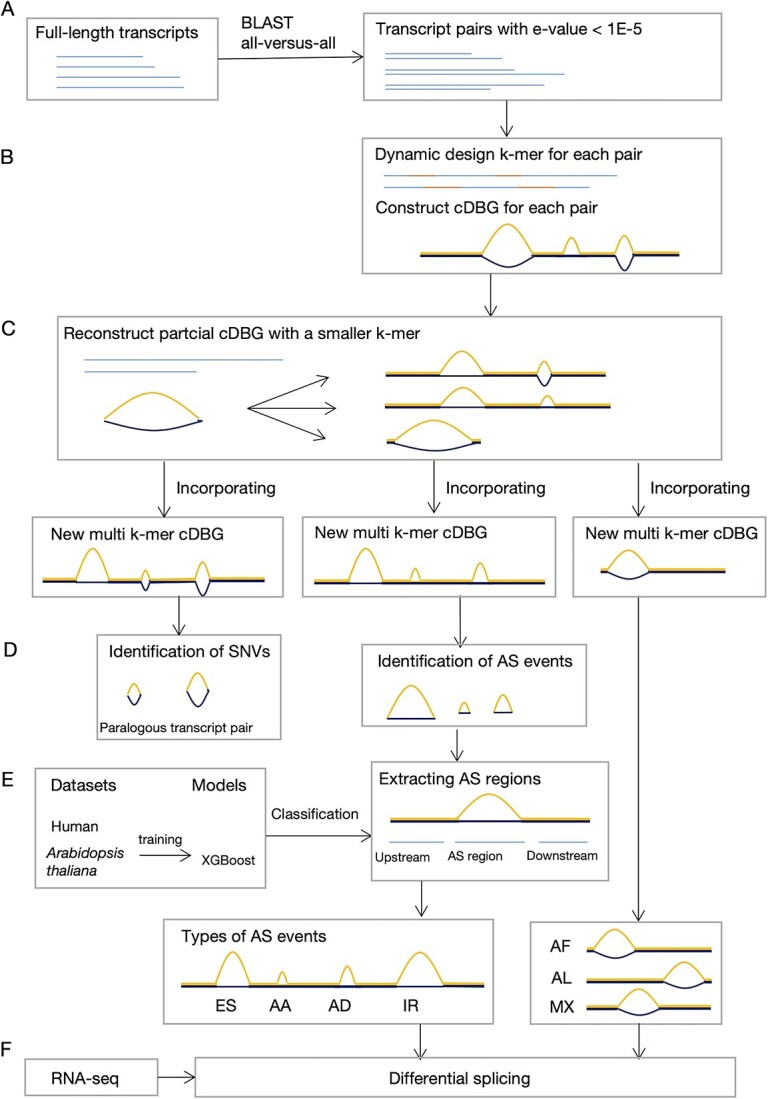
北京师范大学团队提出了一种新的名为“MkcDBGAS”的方法,此方法在没有参考基因组的情况下,只利用转录组来识别所有七种类型的可变剪切事件。
3、Genome Medicine | 发布实现单细胞水平肿瘤细胞群突变与表达异质性的同步研究计算方法
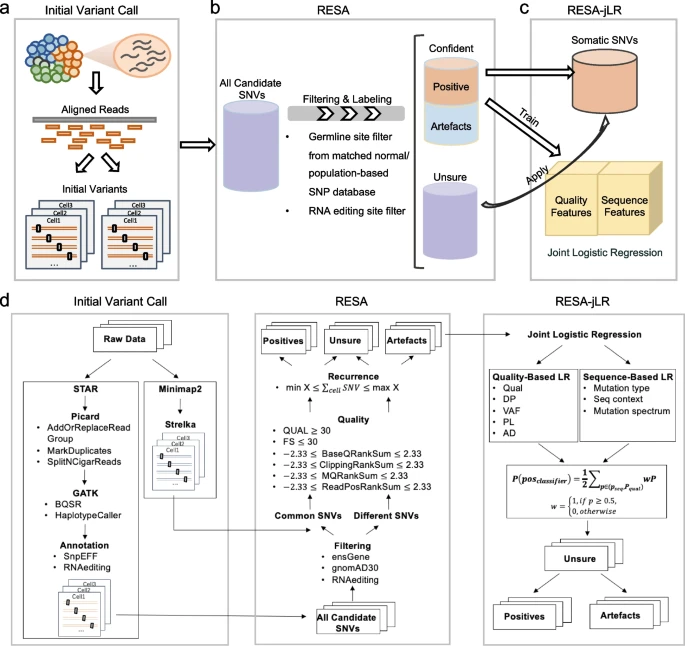
浙江大学报道了一种高精确度(precision)直接从scRNA-seq数据中提取体细胞突变的算法框架RESA(RecurrentlyExpressed SNV Analysis),该算法在多种类型的不同来源的癌症单细胞数据集上进行测试,均取得了较高的精确度。该算法可以应用于癌症的肿瘤异质性与耐药机制研究,实现单细胞水平表达量变化与体细胞突变的双重检测,推进肿瘤单细胞水平基因型到表型变化的理解与认知,对肿瘤分子机制研究、临床诊断、以及个性化治疗方案的制定都有重要意义。
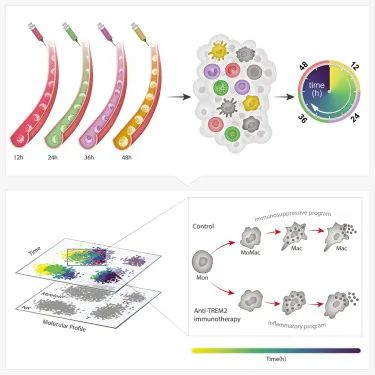
研究人员首次开发出一种跟踪和测量体内单个细胞随时间变化的方法。这种方法被称为zman -seq(来自希伯来语zman,意思是“时间”),这种方法有不同的“时间戳”标记细胞,并在健康或病理组织中跟踪它们。利用这种细胞时间机器,研究人员可以了解细胞的历史,以及每个细胞在组织中停留了多长时间,最终了解组织中发生的分子和细胞的时间变化。

空间转录组测序主要包括5个步骤,包括下游分析部分:空转数据分析和可视化。这篇文章主要分享如何使用python和R读取空转数据,主要使用scanpy stlearn seurat包。

这篇文章总结了在过去的几十年中发生在生物信息学的诸多里程碑,包括早期的蛋白质序列数据库和序列比对方法,到目前的基因组学和新兴的人工智能算法,这些重要的里程碑使我们对生物学的理解产生了重要影响。
7、博文资讯 | 预后模型 vs. 预测模型:别在错误方向上拼尽全力
本文澄清了两个概念:prognostic(预后的)和 predictive(预测的)。
8、博文资讯 | 大海哥带你解码bulk RNA-seq的反卷积新工具 —— BayesPrism
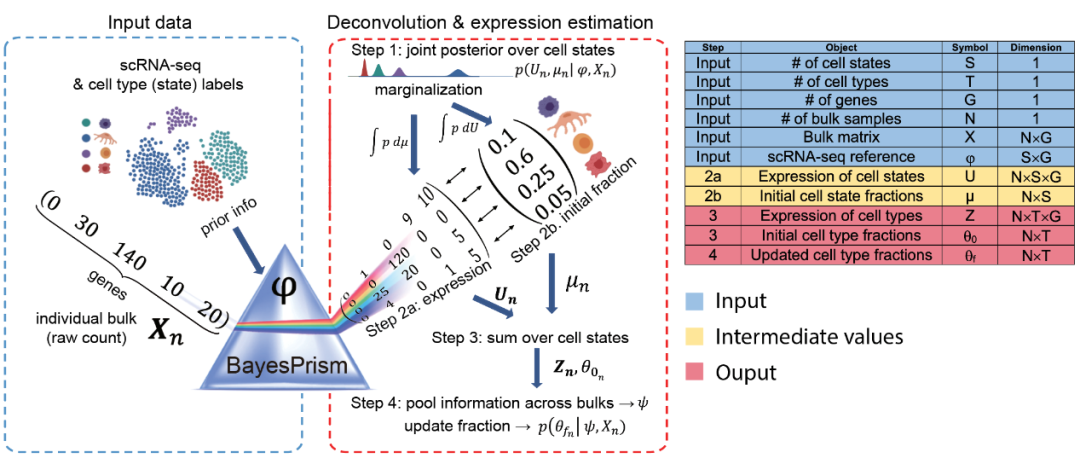
本文介绍了BayesPrism,一种基于贝叶斯模型的前沿技术,它巧妙地将单细胞RNA测序(scRNA-seq)作为参考谱融入Bulk RNA测序(RNA-seq)数据中,从而实现细胞类型评分和基因表达的后验分布推断。
9、hoelzer-lab/rnaflow | 一个基于 Nextflow 的 RNA 差异表达分析流程
##下载整个Bioproject
kingfisher get -p PRJNA486534 -m ena-ascp ena-ftp prefetch aws-http 1>down_prjan486534.log 2>&1
##下载单个样本
kingfisher get -r SRR14615558 -m ena-ascp ena-ftp prefetch aws-http --download-threads 10 1>down.log 2>&1
##下载多个样本
kingfisher get --run-identifiers-list SRR_list.csv -m ena-ascp ena-ftp prefetch --download-threads 10 --check-md5sums 1>down_srr_list.log 2>&1
-p :批量下载BioProject IDs 中的所有数据
-m :指定下载源 ena-ascp、ena-ftp、prefetch、aws-http、aws-cp、gcp-cp 等
-r :下载某个确定的SRA数据
--run-identifiers-list :SRR样本列表文件,单列SRR号
--download-threads -t : 指定线程数
kingfisher是由昆士兰科技大学微生物组研究中心的 Ben J. Woodcroft 教授开发的一款专门用于高通量测序数据下载的工具。其可以用于从公共数据库(ENA、NCBI、SRA、Amazon AWS 和 Google Cloud)获取序列文件及其元数据注释。其输入可以是一个或多个“Run" accession(例如DRR001970),或一个 BioProject accessions(例如PRJNA621514或SRP260223)。
11、理解P值
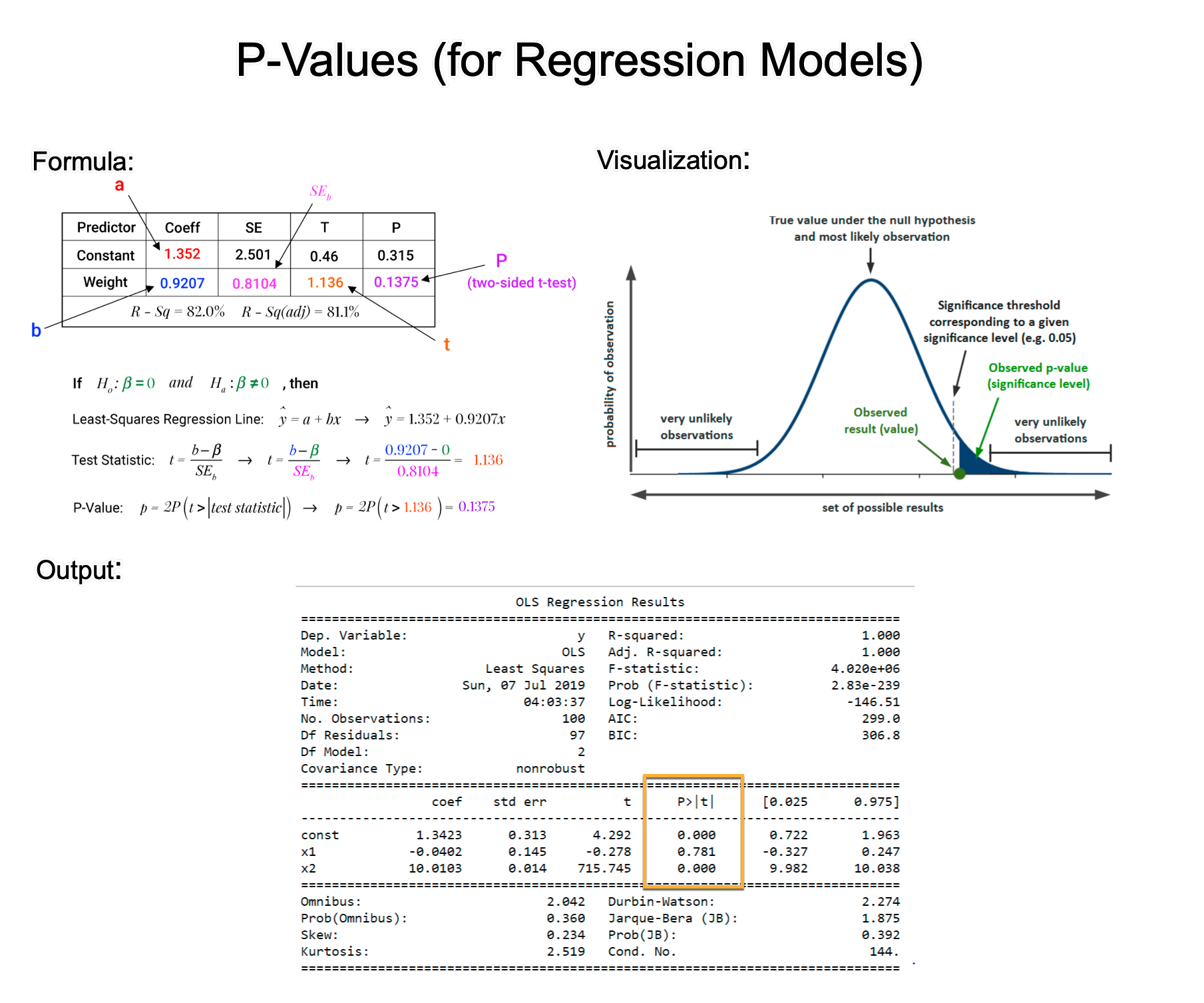
本文用浅显易懂的方式来讲解P值。

本文系统地讲述寻找博士后position需要考虑的问题。
13、史上最好的博士论文
本文分享自Dr. Andrew Stapleton(昵称Andy)斯台普顿博士的博文“The best PhD thesis and dissertations in history”(世上最好的博士论文)。列出了历史上重要的博士论文。
14、Overview of R Modelling Packages

本文概述了用于拟合不同类型回归模型的 R 软件包和函数。
一个基于R学习应用统计的教程,包含代码和讲解。
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)@kongjianyang(空间阳)
这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。

(完)