- Installation
- MaSIF-site
- MaSIF-search
- MaSIF-PDL1-benchmark
- MaSIF-ligand
- Building Docker MaSIF image from a Dockerfile
docker pull pablogainza/masif:latest
docker run -it pablogainza/masif
You now start a local container with MaSIF. The first step should be to update the repository to make sure you have the latest version (in case the image has not been update):
root@b30c52bcb86f:/masif# git pull
Go into the MaSIF site data directory.
root@b30c52bcb86f:/masif# cd data/masif_site/
root@b30c52bcb86f:/masif/data/masif_site#
We will now run MaSIF site on chain A of PDB id 4ZQK. It is important to always input a chain and a PDB id. The first step consists of preparing the data and it is the slowest part of the process. It consists of downloading the pdb, extracting the chain, protonating it, computing the molecular surface and PB electrostatics, and decomposing the protein into patches (about 1 minute on a 120 residue protein):
root@b30c52bcb86f:/masif/data/masif_site# ./data_prepare_one.sh 4ZQK_A
Downloading PDB structure '4ZQK'...
Removing degenerated triangles
Removing degenerated triangles
4ZQK_A
Reading data from input ply surface files.
Dijkstra took 2.28s
Only MDS time: 18.26s
Full loop time: 28.54s
MDS took 28.54s
If you want to run a prediction on multiple chains (e.g. a multidomain protein) you can do so by concatenting all chains (e.g., 4ZQK_AB). You can also run this command on a specific file (i.e. not on a downloaded file) using the --file flag:
root@b30c52bcb86f:/masif/data/masif_site# ./data_prepare_one.sh --file /path/to/myfile/4ZQK.pdb 4ZQK_A
The next step consists of actually running the protein through the neural network to predict interaction sites:
root@b30c52bcb86f:/masif/data/masif_site# ./predict_site.sh 4ZQK_A
Setting model_dir to nn_models/all_feat_3l/model_data/
Setting feat_mask to [1.0, 1.0, 1.0, 1.0, 1.0]
Setting n_conv_layers to 3
Setting out_pred_dir to output/all_feat_3l/pred_data/
Setting out_surf_dir to output/all_feat_3l/pred_surfaces/
(12, 2)
...
Total number of patches for which scores were computed: 2336
GPU time (real time, not actual GPU time): 1.890s
After this step you can find the predictions in numpy files:
root@b30c52bcb86f:/masif/data/masif_site# ls output/all_feat_3l/pred_data
pred_4ZQK_A.npy
root@b30c52bcb86f:/masif/data/masif_site/#
Finally you can run a command to output a ply file with the predicted interface for visualization. A ROC AUC is also computed, but it is only accurate if the protein was found in the original complex (the ground truth is extracted from there):
root@b30c52bcb86f:/masif/data/masif_site# ./color_site.sh 4ZQK_A
Setting model_dir to nn_models/all_feat_3l/model_data/
Setting feat_mask to [1.0, 1.0, 1.0, 1.0, 1.0]
Setting n_conv_layers to 3
Setting out_pred_dir to output/all_feat_3l/pred_data/
Setting out_surf_dir to output/all_feat_3l/pred_surfaces/
ROC AUC score for protein 4ZQK_A : 0.91
Saving output/all_feat_3l/pred_surfaces/4ZQK_A.ply
Computed 1 proteins
Median ROC AUC score: 0.9137235650273854
root@b30c52bcb86f:/masif/data/masif_site#
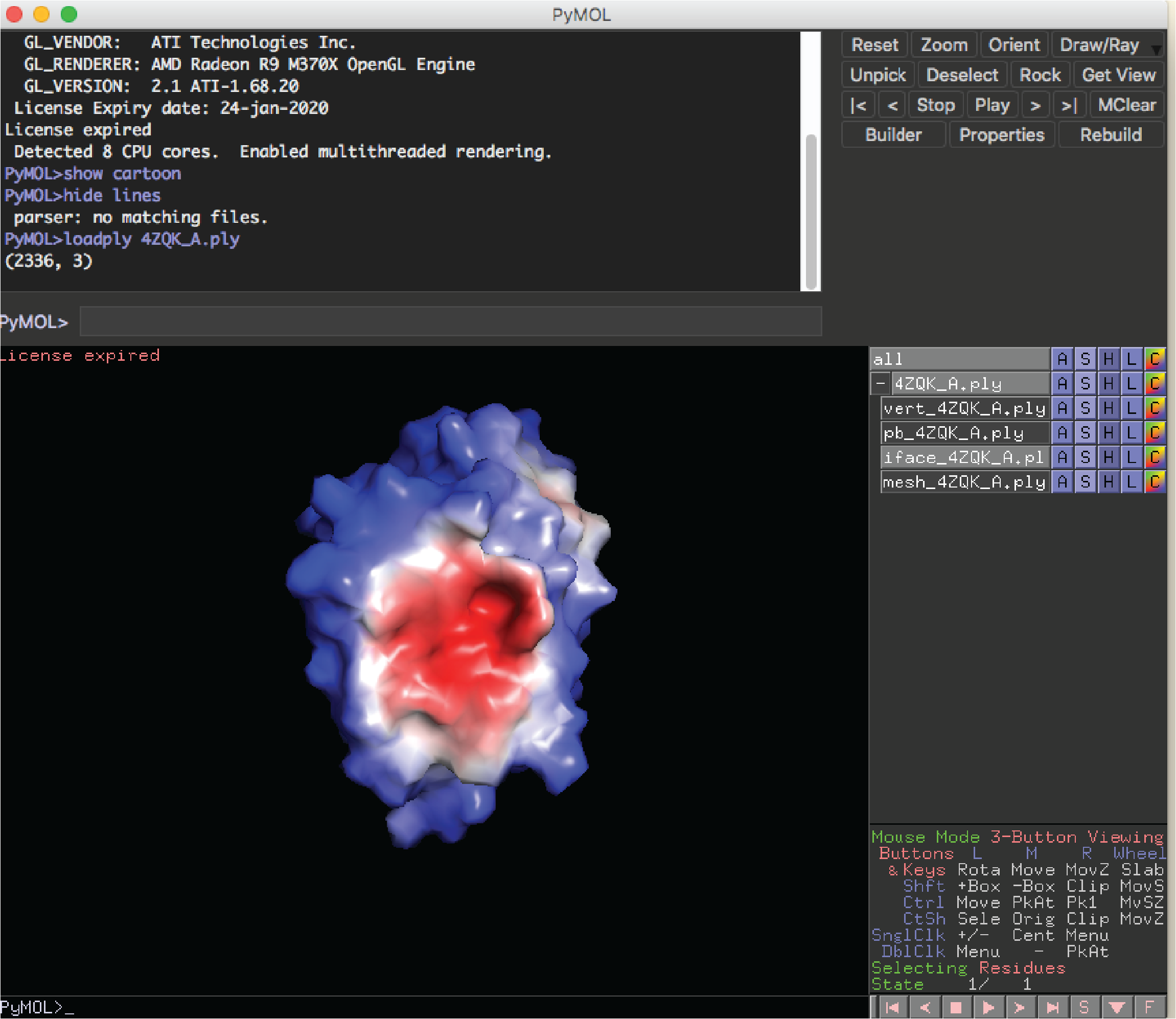
If you have installed the pymol plugin for MaSIF, you can now visualize the predictions. From your local computer run:
docker cp b30c52bcb86f:/masif/data/masif_site/output/all_feat_3l/pred_surfaces/4ZQK_A.ply .
pymol
Then from the pymol command window run:
loadply 4ZQK_A.ply
Then deactivate all objects except the one with 'iface' as part of its name. You should see something like this:
All the MaSIF-site experiments from the paper should be reproducible using the Docker container. For convenience, I have provided a script to reproduce the transient PPI interaction prediction benchmark, which is the one that is compared to state-of-the-art tools (SPPIDER, PSIVER).
cd data/masif_site
./reproduce_transient_benchmark.sh
This process takes about 2 hours, since there are ~60 proteins and they take about 2 minutes to run per protein.
In order to retrain the neural network from zero, I strongly recommend using a cluster to precompute the data and a GPU to train. It will take about 5 days in a single CPU to preprocess all the data. Ideally, one would instead use a cluster. However, if a cluster is not available you can precompute all data by running the commands:
cd data/masif_site
./data_prepare_all.sh
Then, one can train the neural network:
./train_nn.sh
Please make sure to use a Docker version that supports GPU access. You may have to install tensorflow with support for GPU within the Docker image.
This section describes the fast docking benchmark presented in our paper (Table 1).
The fastest way to reproduce this benchmark is to download all the precomputed data from the following site: https://www.dropbox.com/s/09fwtic1095z9z6/masif_ppi_search_precomputed_data.tar.gz?dl=0
Run the masif container and download the data:
docker run -it pablogainza/masif
cd data/masif_ppi_search/
wget https://www.dropbox.com/s/09fwtic1095z9z6/masif_ppi_search_precomputed_data.tar.gz?dl=0
tar cvfz masif_ppi_search_precomputed_data.tar.gz
Change the directory to the benchmark directory and run the benchmark for a number of decoys K (e.g. 100 or 2000 as in the paper):
cd ../../comparison/masif_ppi_search/masif_descriptors_nn/
./second_stage_masif.sh 100
The results should be the last line of the results_masif.txt file.
If you wish, you can also reproduce the benchmark data. I have conveniently left a script to recompute the data:
docker run -it pablogainza/masif
cd data/masif_ppi_search/
././recompute_data_docking_benchmark.sh
This should take about 3 minutes per protein on a CPU (about 2 on a GPU). For a total of 100 protein pairs it may take a few hours.
Finally change the directory to the benchmark directory and run the benchmark for a number of decoys K (e.g. 100 or 2000 as in the paper):
cd ../../comparison/masif_ppi_search/masif_descriptors_nn/
./second_stage_masif.sh 100
For this task I strongly recommend a cluster to do the precomputation because there are about 10000 proteins per cluster. The steps to recompute and retrain are laid out in the main MaSIF readme.
Similar as for the bound:
docker run -it pablogainza/masif
cd data/masif_ppi_search_ub/
wget https://www.dropbox.com/s/5w46ankuk3y2edo/masif_ppi_search_ub_precomputed_data.tar.gz?dl=0
tar cvfz masif_ppi_search_ub_precomputed_data.tar.gz
Change the directory to the benchmark directory and run the benchmark for a number of decoys K (e.g. 2000 as in the paper):
cd ../../comparison/masif_ppi_search_ub/masif_descriptors_nn/
./second_stage_masif.sh 2000
If you wish, you can also reproduce the benchmark data. I have conveniently left a script to recompute the data:
docker run -it pablogainza/masif
cd data/masif_ppi_search_ub/
./recompute_data_docking_benchmark.sh
This should take about 3 minutes per protein on a CPU (about 2 on a GPU). For a total of 40 protein pairs it may take a few hours.
Finally change the directory to the benchmark directory and run the benchmark for a number of decoys K (e.g. 2000 as in the paper):
cd ../../comparison/masif_ppi_search_ub/masif_descriptors_nn/
./second_stage_masif.sh 2000
In the paper we present a benchmark to scan ~11000 proteins for the binder of PD-L1 (taken from the co-crystal structure). This benchmark is very fast - finishes in minutes. The benchmark works as follows:
(a) First, based on the MaSIF-site predictions, the center of the interface for PD-L1 is chosen.
(b) Then, the fingerprint for that point is matched to the fingerprints o ftens of millions of patches from the database of 11000 proteins, and those that are within a cutoff are selected for further processing.
(c) each patch that passes the fingerprint is aligned and scored with a neural network.
For convenience, I have uploaded all the preprocessed data to Dropbox (eventually this will be replaced by a Zenodo link): https://www.dropbox.com/s/aaf5nt6smbrx8p7/masif_pdl1_benchmark_precomputed_data.tar?dl=0
Steps to reproduce the benchmark.
Download the compressed data files to your local machine and unpack. You must make a temporary directory in your host machine to download a large file (about 30GB) which will contain the benchmark data. Here this directory is called '/your/temporary/path/docker_files/'.
mkdir /your/temporary/path/docker_files/
wget https://www.dropbox.com/s/aaf5nt6smbrx8p7/masif_pdl1_benchmark_precomputed_data.tar?dl=0
tar xvf masif_pdl1_benchmark_precomputed_data.tar
rm masif_pdl1_benchmark_precomputed_data.tar
You should now have a list of compressed tar.gz files.
Start the docker container for masif, linking the directory in your host machine.
docker run -it -v /your/temporary/path/docker_files/:/var/docker_files/ pablogainza/masif
Pull the latest version from the masif repository
root@b30c52bcb86f:/masif# git pull
Go into the pdl1 benchmark data directory and untar all the downloaded data files:
cd data/masif_pdl1_benchmark/
tar xvfz /var/docker_files/4ZQK_p1_desc_flipped.tar.gz -C .
tar xvfz /var/docker_files/4ZQK_surf_pred.tar.gz -C .
tar xvfz /var/docker_files/list_indices.tar.gz -C .
tar xvfz /var/docker_files/masif_search_descriptors.tar.gz -C .
tar xvfz /var/docker_files/masif_site_predictions.tar.gz -C .
tar xvfz /var/docker_files/pdbs.tar.gz -C .
tar xvfz /var/docker_files/plys.tar.gz -C .
The -C flag force the unpacking to occur in the current directory. Finally run the benchmark.
./run_benchmark_nn.sh
This takes some time to run (~30 minutes). After this you can sort scores:
cat log.txt | sort -k 2 -n
You can also visualize the top candidates who were all stored in the out/ directory.
*** A note on descriptors distance *** A critical value now is the cutoff used for masif-search's fingerprint distance. In general, and as explained in the paper, the lower the cutoff, the less the number of results, and therefore the faster the run. By default, the value is set here at 1.7, which works well for this dataset. However, it may be possible that you need to relax this further (to, say, 2.0 or 2.2). You can try different values.
You can run this protocol on your protein of interest as well. In general, for it to work you need a target with a high shape complementarity, and one in which MaSIF correctly labels the site. You probably may also have to play with the descriptor distance parameters.
This tutorial will be soon available
To build a Docker MaSIF image from a Dockerfile run the following steps:
git clone https://github.com/LPDI-EPFL/masif-dockerfile
docker build -t masif_docker .