[Alignment] Multiple Sequence Alignment (MSA) and Alignment Graph #1763
Comments
|
Hello @cartoonist! We unfortunately haven't implemented a multiple sequence alignment, yet. But, we already had some discussions how to implement it in seqan3 and we definitely want to implement this feature (we also have some projects that need them). Since you already have experience with seqan2, maybe you could help us design this feature by answering these questions:
Thank you! |
|
Thanks for your prompt reply. I am currently looking for a way to get a "partial-order graph" representing MSA of a set of sequences. I came across seqan2 implementation of T-Coffee algorithm resulting an For implementing it in seqan2, I mostly followed the documentation to call using string_type = seqan::String< seqan::AminoAcid >;
using stringset_type = seqan::StringSet< string_type >;
using dependentset_type = seqan::StringSet< string_type, seqan::Dependent<> >;
using alignment_type = seqan::Alignment< dependentset_type, void >;
using graph_type = seqan::Graph< alignment_type >;
auto scoring_scheme = seqan::Blosum62( -1, -11 );
stringset_type sequences;
//dependentset_type dsequences;
while ( fasta_file >> record ) {
appendValue( sequences, record.seq );
//appendValue( dsequences, back( sequences ) );
}
graph_type align_graph( sequences );
//graph_type align_graph( dsequences );
globalMsaAlignment( align_graph, scoring_scheme );The only part that I found user-unfriendly is that the graph type is only defined for the Since I don't know about implementation details, I might be mistaken. But what I expect here is that the graph type should be defined for all string set types. In order to avoid unnecessary copy, it can internally keep a dependent string set initialised by the given string set which can be either a dependent or an owner. Regarding the last point, I realised that seqan3 is quite different in design. Since I have not worked with it, I don't have an idea how the interface should look like in seqan3. However, broadly speaking, I prefer those APIs close to the standard library, such as sdsl-lite. For example, heavy use of interface functions makes the library hard to navigate and, consequently, hard to extend because one has to look for the overloaded functions through lots of header files to figure out what is happening under the hood. I personally prefer member functions for basic operations such as I hope I could answer your points. |
|
Thank you for the snippet, can you show me how you process the constructed Just some answers from the points that I skimmed over:
|
|
Hello @cartoonist, good news! We started to work on a design proposal for multiple sequence alignment (seqan/product_backlog#104). Since we would like to include your use case, it would be awesome if you could answer my last questions:
Best regards |
|
Hi @cartoonist, it would be awesome if you could take a couple of minutes and answer my questions. Thank you! |
|
Hello @marehr, Really sorry for my late response! I wanted to get back to you with a concrete example/snippet. But for now, please let me update you with my current objective/use case, later I will post some examples or snippets if needed. I would like to create a pan-gnome graph from a multiple sequence alignment without flattening the MSA; e.g. dumping it to a FASTA file. This graph itself can be used independently or can be embedded in a larger graph (e.g. a whole genome graph). I still could not figure out how to construct a partial order graph using in seqan2. I did try to visualise the alignment graph constructed by seqan. It seems that nodes corresponding to alignment segments are connect together and the resulting graph is a forest of complete graphs. That was different from what I expected as a partial order graph. Right now, as temporary workaround, I just dump the MSA in FASTA format then use an external Python script converting FASTA to GFA. Assume that I have a partial order graph, what I need is, external or internal ( P.S. Thanks for the pointers to seqan3 in your previous reply. |
|
Awesome!
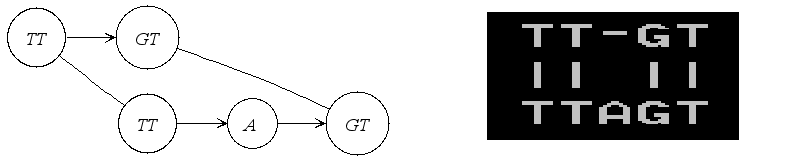
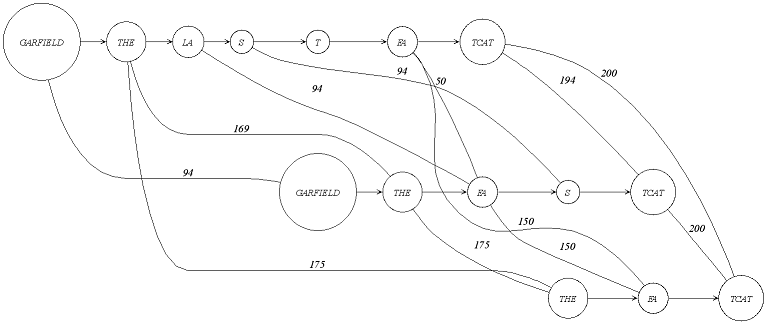
We have those two images in our seqan2 docs
and
From what I understood the "alignment graph" stores all shared text segments (according to the alignment) in a node and has multiple edges to nodes (It compresses nodes of characters into nodes of sub-sequences). I never worked with it, so I'm not to sure of the internal representation. Does this resemble your partial order graph, or do you have some drawing of 2 / 3 sequences that depicts your use case?
You say "ideally", does this mean that all enumerated sub-paths would suffice for you, or do you want that as a convenience function that gives you the sub-sequence up until that node? |
|
AFAIK, the graph is a partially ordered with respect to a sequence. This is resembled by the directed edges connecting nodes one one level (representing one sequence). In addition, you have the undirected edges connecting nodes (sequence segments) between the sequences (IIRC than PO graph would make them to one node, right?). Our graph is kind of a generalisation where one node represents exactly one character. Our SeqAn2 graph implementation offers all kind of traversals. So I think it should be possible to do what you want. But I have to be honest, I am also not familiar with the interfaces and would need to dig deeper to figure out how you can traverse along specific paths. Here is a tutorial that might offer some help with this for now. https://seqan.readthedocs.io/en/develop/Tutorial/DataStructures/Graphs.html#graph-iterators. Another interesting topic that might be relevant is that there was a publication that describes how the alignment graph can be converted into another pan-genome graph representation. https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-99 Not sure if you knew this already, but maybe that also offers some more information. Then we could use these insights to derive the design for the new implementation. I think, pan-genome analysis together with our work on compressive genomics will be quite the nice addition to our library. |
|
Hi, I am also interested in Partial Order Alignment, or just progressive sequence to graph alignment. For the current version of Seqan3, is the path information stored for each sequence aligned to the graph? Of course it is possible to traverse the graph and chose any arbitrary path that matches the sequence ad hoc, but because POA is order dependent (aka progressive or "greedy"), there can be cases where the addition of branches in the graph can change successive alignments of the same sequence. This means the ad hoc method is not sufficient to find the actual path an alignment took. Alternatively, if any MSA algorithm can be converted to a graph with path information, that would be nice too. |
|
We are also tracking MSA design decisions here: seqan/product_backlog#104 |
Question
I have just started using seqan3. I have worked with seqan2 though. I want to ask about multiple sequence alignment and alignment graphs in seqan3. I could not find anything about MSA in the documentation. Are they missing in the documentation or they are not implemented yet?
The text was updated successfully, but these errors were encountered: