Before opening a new issue here, please check the appropriate help channel on the KneadData bioBakery Support Forum and consider opening or commenting on a thread there.
For additional information, visit the KneadData Tutorial.
KneadData is a tool designed to perform quality control on metagenomic and metatranscriptomic sequencing data, especially data from microbiome experiments. In these experiments, samples are typically taken from a host in hopes of learning something about the microbial community on the host. However, sequencing data from such experiments will often contain a high ratio of host to bacterial reads. This tool aims to perform principled in silico separation of bacterial reads from these "contaminant" reads, be they from the host, from bacterial 16S sequences, or other user-defined sources. Additionally, KneadData can be used for other filtering tasks. For example, if one is trying to clean data derived from a human sequencing experiment, KneadData can be used to separate the human and the non-human reads.
If you use the KneadData software, please cite our manuscript: TBA
- Trimmomatic (version == 0.33) (automatically installed)
- Bowtie2 (version >= 2.2) (automatically installed)
- Python (version >= 2.7)
- Java Runtime Environment
- TRF (optional)
- Fastqc (optional)
- SAMTools (only required if input file is in BAM format)
- Memory (>= 4 Gb if using Bowtie2, >= 8 Gb if using BMTagger)
- Operating system (Linux or Mac)
Optionally, BMTagger can be used instead of Bowtie2.
The executables for the required software packages should be installed in your $PATH. Alternatively, you can provide the location of the Bowtie2 install ($BOWTIE2_DIR) with the following KneadData option “--bowtie2 $BOWTIE2_DIR”.
Before installing KneadData, please install the Java Runtime Environment (JRE). First download the JRE for your platform. Then follow the instructions for your platform: Linux 64-bit or Mac OS. At the end of the installation, add the location of the java executable to your $PATH.
You can download the latest KneadData release or the development version. The source contains example files. If installing with pip, it is optional to first download the KneadData source.
Option 1: Latest Release (Recommended)
- Download kneaddata.tar.gz and unpack the latest release of KneadData.
Option 2: Development Version
-
Create a clone of the repository:
$ git clone https://github.com/biobakery/kneaddata.gitNote: Creating a clone of the repository requires Git to be installed.
$ pip install kneaddata- This command will automatically install Trimmomatic and Bowtie2. To bypass the install of dependencies, add the option "--install-option='--bypass-dependencies-install'".
- If you do not have write permissions to '/usr/lib/', then add the option "--user" to the install command. This will install the python package into subdirectories of '$HOME/.local'. Please note when using the "--user" install option on some platforms, you might need to add '$HOME/.local/bin/' to your $PATH as it might not be included by default. You will know if it needs to be added if you see the following message
kneaddata: command not foundwhen trying to run KneadData after installing with the "--user" option.
- Follow the instructions to download KneadData
- Move to the KneadData source directory:
$ cd kneaddata - Install KneadData
$ python setup.py install- This command will automatically install Trimmomatic and Bowtie2. To bypass the install of dependencies, add the option "--bypass-dependencies-install".
- If you do not have write permissions to '/usr/lib/', then add the option "--user" to the install command. This will install the python package into subdirectories of '$HOME/.local'. Please note when using the "--user" install option on some platforms, you might need to add '$HOME/.local/bin/' to your $PATH as it might not be included by default. You will know if it needs to be added if you see the following message
kneaddata: command not foundwhen trying to run KneadData after installing with the "--user" option.
It is recommended that you download the human (hg37_and_human_contamination) reference database (approx. size = 3.5 GB). However, this step is not required if you are using your own custom reference database or if you will not be running with a reference database.
This database is based on the Decoy Genome (http://www.cureffi.org/2013/02/01/the-decoy-genome/) and contaminants taken from “Human contamination in bacterial genomes has created thousands of spurious proteins” (Salzberg et. al. 2019)
$ kneaddata_database --download human_genome bowtie2 $DIR- When running this command, $DIR should be replaced with the full path to the directory you have selected to store the database.
If you are running with bmtagger instead of bowtie2, then download the bmtagger database instead of the bowtie2 database with the following command.
$ kneaddata_database --download human_genome bmtagger $DIR- When running this command, $DIR should be replaced with the full path to the directory you have selected to store the database.
The human transcriptome (hg38) reference database is also available for download (approx. size = 254 MB).
$ kneaddata_database --download human_transcriptome bowtie2 $DIR
The SILVA Ribosomal RNA reference database is also available for download (approx. size = 11 GB).
$ kneaddata_database --download ribosomal_RNA bowtie2 $DIR
The mouse (C57BL) reference database is also available for download (approx. size = 3 GB).
$ kneaddata_database --download mouse_C57BL bowtie2 $DIR
A reference database can be downloaded to use when running KneadData. Alternatively, you can create your own custom reference database.
First you must select reference sequences for the contamination you are trying to remove. Say you wish to filter reads from a particular "host." Broadly defined, the host can be an organism, or a set of organisms, or just a set of sequences. Then, you simply must generate a reference database for KneadData from a FASTA file containing these sequences. Usually, researchers want to remove reads from the human genome, the human transcriptome, or ribosomal RNA. You can access some of these FASTA files using the resources below:
-
Ribosomal RNA: Silva provides a comprehensive database for ribosomal RNA sequences spanning all three domains of life (Bacteria, Archaea, and Eukarya).
-
Human Genome & Transcriptome: Information about the newest assembly of human genomic data can be found at the NCBI project page. USCS provides a convenient website to download this data.
KneadData requires that your reference sequences (FASTA files) be indexed to form KneadData databases beforehand. This only needs to be done once per reference sequence.
For certain common databases, we provide indexed files. If you use these, you can skip the manual build steps below. Alternatively if you would like to bypass the reference alignment portion of the workflow, a database does not need to be provided when running KneadData.
To download the indexed human reference database, run the following command:
$ kneaddata_database --download human bowtie2 $DIR- When running this command, $DIR should be replaced with the full path to the directory you have selected to store the database.
Simply run the bowtie2-build indexer included with Bowtie2 as follows:
$ bowtie2-build <reference> <db-name>
Where <reference> is the reference FASTA file, and <db-name> is the name you
wish to call your Bowtie2 database. For more details, refer
to the bowtie2-build-documentation
Creating the SILVA ribosomal_RNA database requires one additional step. Run the following python program before bowtie2-build command which converts the "U"s to "T"s in the fasta sequences.
Script link: modify_RNA_to_DNA.py
$ python -u modify_RNA_to_DNA.py input.fasta output.fa
KneadData includes kneaddata_build_database, an executable that
will automatically generate these databases for BMTagger. Simply run
$ kneaddata_build_database reference.fasta
By default, this will generate the reference databases, whose names are prefixed
with reference.fasta.
A note on PATH: The above command will fail if the tools in the BMTagger suite
(specifically, bmtool and srprism) and the NCBI BLAST executables are not in
your PATH. If this is the case, you can specify a path to these tools using the
-b, -s, and -m options. Run
$ kneaddata_build_database --help
for more details.
Say you want to remove human reads from your metagenomic sequencing data.
You downloaded the human genome in a file called Homo_sapiens.fasta.
Then, you can generate the KneadData database by executing:
$ bowtie2-build Homo_sapiens.fasta -o Homo_sapiens_db
for Bowtie2, or
$ kneaddata_build_database Homo_sapiens.fasta -o Homo_sapiens_db
All of the required KneadData database files will have file names prefixed by
Homo_sapiens_db and have various file extensions.
Run the following python program before bowtie2-build command which converts the "U"s to "T"s in the fasta sequences for creating SILVA ribosomal_RNA database.
Script link: modify_RNA_to_DNA.py
$ python -u modify_RNA_to_DNA.py input.fasta output.fa
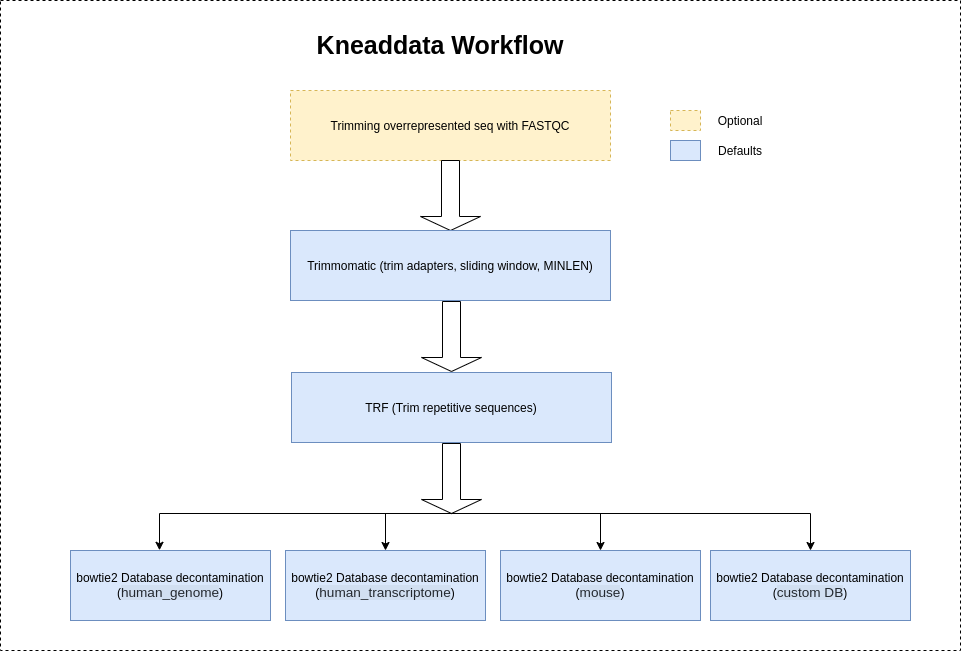
After downloading or generating your database file, you can start to remove contaminant reads. As input, KneadData requires FASTQ files. It supports both single end and paired end reads. KneadData uses either Bowtie2 (default) or BMTagger to identify the contaminant reads.
To run KneadData in single end mode, run
$ kneaddata --unpaired seq.fastq --reference-db $DATABASE --output kneaddata_output
This will create files in the folder kneaddata_output named
seq_kneaddata_$DATABASE_bowtie2_contam.fastq: FASTQ file containing reads that were identified as contaminants from the database (named $DATABASE).seq_kneaddata.fastq: This file includes reads that were not in the reference database.seq_kneaddata.trimmed.fastq: This file has trimmed reads.seq_kneaddata.log
To run KneadData in single end mode with BMTagger, run
$ kneaddata --unpaired seq.fastq --reference-db $DATABASE --run-bmtagger
By default, this will create the same four files as running with bowtie2. The only differences are the contaminants file will have "bmtagger" in the name instead of "bowtie2" and the included $DATABASE name would differ.
If you wanted to use BMTagger and the BMTagger executable was located at
$HOME/bmtagger/bmtagger.sh which is not in your $PATH you would add the option "--bmtagger $HOME/bmtagger/bmtagger.sh" to the command.
If you wanted to select the basenames of the output files, you would add the option "--output-prefix $NAME", replacing $NAME with the name you would like used.
To run KneadData in paired end mode with Bowtie2, run
$ kneaddata --input1 seq1.fastq --input2 seq2.fastq -db $DATABASE --output kneaddata_output
To run KneadData in paired end mode with BMTagger, run
$ kneaddata --input seq1.fastq --input seq2.fastq -db $DATABASE --run-bmtagger --output kneaddata_output
seq1.fastq: Your input FASTQ file, first mateseq2.fastq: Your input FASTQ file, second mate$DATABASE: Prefix for the KneadData database.kneaddata_output: The folder to write the output files.
The outputs depend on what happens during the quality filtering and trimming part of the pipeline.
When performing quality filtering and trimming for paired end files, three things can happen:
- Both reads in the pair pass.
- The read in the first mate passes, and the one in the second does not pass.
- The read in the second mate passes, and the one in the first does not pass.
The number of outputs are a function of the read quality.
KneadData + Bowtie2 (or BMTagger) Outputs: There can be up to 8 outputs per reference database, plus up to 5 aggregate outputs.
Instead of single end reads, say you have paired end reads and you want to
separate the reads that came from bacterial mRNA, bacterial rRNA, and human RNA.
You have two databases, one prefixed bact_rrna_db and the other prefixed
human_rna_db, and your sequence files are seq1.fastq and seq2.fastq. To
run with Bowtie2, execute
$ kneaddata --input1 seq1.fastq --input2 seq2.fastq -db bact_rrna_db -db human_rna_db --output seq_out
This will output files in the folder seq_out named:
Files for just the bact_rrna_db database:
seq_kneaddata_paired_bact_rrna_db_bowtie2_contam_1.fastq: Reads from the first mate in situation (1) above that were identified as belonging to thebact_rrna_dbdatabase.seq_kneaddata_paired_bact_rrna_db_bowtie2_contam_2.fastq: Reads from the second mate in situation (1) above that were identified as belonging to thebact_rrna_dbdatabase.seq_kneaddata_paired_bact_rrna_db_bowtie2_clean_1.fastq: Reads from the first mate in situation (1) above that were identified as NOT belonging to thebact_rrna_dbdatabase.seq_kneaddata_paired_bact_rrna_db_bowtie2_clean_2.fastq: Reads from the second mate in situation (1) above that were identified as NOT belonging to thebact_rrna_dbdatabase.
Depending on the input FASTQ, one or more of the following may be output:
seq_kneaddata_unmatched_1_bact_rrna_db_bowtie2_contam.fastq: Reads from the first mate in situation (2) above that were identified as belonging to thebact_rrna_dbdatabase.seq_kneaddata_unmatched_1_bact_rrna_db_bowtie2_clean.fastq: Reads from the first mate in situation (2) above that were identified as NOT belonging to thebact_rrna_dbdatabase.seq_kneaddata_unmatched_2_bact_rrna_db_bowtie2_contam.fastq: Reads from the second mate in situation (3) above that were identified as belonging to thebact_rrna_dbdatabase.seq_kneaddata_unmatched_2_bact_rrna_db_bowtie2_clean.fastq: Reads from the second mate in situation (3) above that were identified as NOT belonging to thebact_rrna_dbdatabase.
Files for just the human_rna_db database:
seq_kneaddata_paired_human_rna_db_bowtie2_contam_1.fastq: Reads from the first mate in situation (1) above that were identified as belonging to thehuman_rna_dbdatabase.seq_kneaddata_paired_human_rna_db_bowtie2_contam_2.fastq: Reads from the second mate in situation (1) above that were identified as belonging to thehuman_rna_dbdatabase.seq_kneaddata_paired_human_rna_db_bowtie2_clean_1.fastq: Reads from the first mate in situation (1) above that were identified as NOT belonging to thehuman_rna_dbdatabase.seq_kneaddata_paired_human_rna_db_bowtie2_clean_2.fastq: Reads from the second mate in situation (1) above that were identified as NOT belonging to thehuman_rna_dbdatabase.
Depending on the input FASTQ, one or more of the following may be output:
seq_kneaddata_unmatched_1_human_rna_db_bowtie2_contam.fastq: Reads from the first mate in situation (2) above that were identified as belonging to thehuman_rna_dbdatabase.seq_kneaddata_unmatched_1_human_rna_db_bowtie2_clean.fastq: Reads from the first mate in situation (2) above that were identified as NOT belonging to thehuman_rna_dbdatabase.seq_kneaddata_unmatched_2_human_rna_db_bowtie2_contam.fastq: Reads from the second mate in situation (2) above that were identified as belonging to thehuman_rna_dbdatabase.seq_kneaddata_unmatched_2_human_rna_db_bowtie2_clean.fastq: Reads from the second mate in situation (2) above that were identified as NOT belonging to thehuman_rna_dbdatabase.
Note, the files named "*_clean.fastq" will only be written if running with the option "--store-temp-output".
Aggregated files:
seq_kneaddata.log: Log file containing statistics about the run.seq_kneaddata_paired_1.fastq: Reads from the first mate in situation (1) identified as NOT belonging to any of the reference databases.seq_kneaddata_paired_2.fastq: Reads from the second mate in situation (1) identified as NOT belonging to any of the reference databases.seq_kneaddata_unmatched_1.fastq: Reads from the first mate in situation (2) identified as NOT belonging to any of the reference databases.seq_kneaddata_unmatched_2.fastq: Reads from the second mate in situation (3) identified as NOT belonging to any of the reference databases.
The examples folder contains a demo input file. This file is a single read, fastq format.
$ kneaddata --unpaired examples/demo.fastq --reference-db examples/demo_db --output kneaddata_demo_output
This will create four output files:
kneaddata_demo_output/demo_kneaddata.fastqkneaddata_demo_output/demo_kneaddata_demo_db_bowtie2_contam.fastqkneaddata_demo_output/demo_kneaddata.logkneaddata_demo_output/demo_kneaddata.trimmed.fastq
Kneaddata will use "NexteraPE" adapters provided by trimomatic to trim the adapter contents by default.
The other available options are: ["NexteraPE", "TruSeq2", "TruSeq3","none"]. Based on the source of the sequencer and the FASTQC report, it is highly reccommended
to choose the correct sequencer source to ensure the removal of adapter contents by Kneaddata.
kneaddata --unpaired demo.fastq -db demo_db -o kneaddata_output --sequencer-source TruSeq3 --fastqc FastQC
kneaddata --unpaired demo.fastq -db demo_db -o kneaddata_output --sequencer-source none --fastqc FastQC
It is highly recommeded to use --run-trim-repetitive flag for Shotgun sequences (Metatranscriptomics-MTX, Metagenomics-MGX) to trim the overrepresented sequences if shown in FASTQC reports.
However, Kneaddata will not trim the overrepresented sequences by default as Amplicon sequences usually have a large number of repetitive reads resulting in depletion of the read count.
kneaddata --unpaired demo.fastq -db demo_db -o kneaddata_output --run-trim-repetitive --fastqc FastQC
kneaddata --unpaired demo.fastq -db demo_db -o kneaddata_output --run-trim-repetitive --sequencer-source TruSeq3 --fastqc FastQC
If you want to specify additional arguments for Bowtie2 using the
--bowtie2-options flag, you will need to use the equals sign along with quotes. Add additional flags for each option.
For example:
$ kneaddata --unpaired demo.fastq --output kneaddata_output --reference-db database_folder --bowtie2-options="--very-fast" --bowtie2-options="-p 2"
A similar approach is used to specify additional arguments for Trimmomatic:
$ kneaddata --unpaired demo.fastq --output kneaddata_output --reference-db database_folder --trimmomatic-options="LEADING:3" --trimmomatic-options="TRAILING:3"
NOTE: Manually specifying additional arguments will completely override the defaults.
Also more than one database can be provided for each run. The database argument can contain the folder that includes the database or the prefix of the database files.
For example:
$ kneaddata --unpaired demo.fastq --output kneaddata_output --reference-db database_folder --reference-db database_folder2/demo
Thanks go to these wonderful people:
All options can be accessed with $ kneaddata --help.
usage: kneaddata [-h] [--version] [-v] [-i1 INPUT1] [-i2 INPUT2]
[-un UNPAIRED] -o OUTPUT_DIR
[-db REFERENCE_DB] [--bypass-trim] [--run-trim-repetitive]
[--output-prefix OUTPUT_PREFIX] [-t <1>] [-p <1>]
[-q {phred33,phred64}] [--run-bmtagger]
[--run-fastqc-start] [--run-fastqc-end] [--store-temp-output]
[--cat-final-output]
[--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL}] [--log LOG]
[--trimmomatic TRIMMOMATIC_PATH] [--max-memory MAX_MEMORY]
[--trimmomatic-options TRIMMOMATIC_OPTIONS]
[--bowtie2 BOWTIE2_PATH] [--bowtie2-options BOWTIE2_OPTIONS]
[--bmtagger BMTAGGER_PATH] [--trf TRF_PATH] [--match MATCH]
[--mismatch MISMATCH] [--delta DELTA] [--pm PM] [--pi PI]
[--minscore MINSCORE] [--maxperiod MAXPERIOD]
[--fastqc FASTQC_PATH]
KneadData
optional arguments:
-h, --help show this help message and exit
-v, --verbose additional output is printed
global options:
--version show program's version number and exit
-i INPUT, --input INPUT
input FASTQ file (add a second argument instance to run with paired input files)
-o OUTPUT_DIR, --output OUTPUT_DIR
directory to write output files
-db REFERENCE_DB, --reference-db REFERENCE_DB
location of reference database (additional arguments add databases)
--run-trim-repetitive Option to trim repetitive/overrepresented sequences generated by FASTQC reports
--bypass-trim bypass the trim step
--output-prefix OUTPUT_PREFIX
prefix for all output files
[ DEFAULT : $SAMPLE_kneaddata ]
-t <1>, --threads <1>
number of threads
[ Default : 1 ]
-p <1>, --processes <1>
number of processes
[ Default : 1 ]
-q {phred33,phred64}, --quality-scores {phred33,phred64}
quality scores
[ DEFAULT : phred33 ]
--run-bmtagger run BMTagger instead of Bowtie2 to identify contaminant reads
--bypass-trf option to bypass the removal of tandem repeats
--run-fastqc-start run fastqc at the beginning of the workflow
--run-fastqc-end run fastqc at the end of the workflow
--store-temp-output store temp output files
[ DEFAULT : temp output files are removed ]
--cat-final-output concatenate all final output files
[ DEFAULT : final output is not concatenated ]
--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL}
level of log messages
[ DEFAULT : DEBUG ]
--log LOG log file
[ DEFAULT : $OUTPUT_DIR/$SAMPLE_kneaddata.log ]
trimmomatic arguments:
--trimmomatic TRIMMOMATIC_PATH
path to trimmomatic
[ DEFAULT : $PATH ]
--max-memory MAX_MEMORY
max amount of memory
[ DEFAULT : 500m ]
--trimmomatic-options TRIMMOMATIC_OPTIONS
options for trimmomatic
[ DEFAULT : SLIDINGWINDOW:4:20 MINLEN:50 ]
MINLEN is set to 50 percent of total input read length. The user can alternatively specify a length (in bases) for MINLEN.
--sequencer-source options for sequencer-source
[ DEFAULT: NexteraPE]
Available sequencers: ["NexteraPE","TruSeq2","TruSeq3"]
bowtie2 arguments:
--bowtie2 BOWTIE2_PATH
path to bowtie2
[ DEFAULT : $PATH ]
--bowtie2-options BOWTIE2_OPTIONS
options for bowtie2
[ DEFAULT : --very-sensitive ]
bmtagger arguments:
--bmtagger BMTAGGER_PATH
path to BMTagger
[ DEFAULT : $PATH ]
trf arguments:
--bypass-trf bypass the TRF step
--trf TRF_PATH path to TRF
[ DEFAULT : $PATH ]
--match MATCH matching weight
[ DEFAULT : 2 ]
--mismatch MISMATCH mismatching penalty
[ DEFAULT : 7 ]
--delta DELTA indel penalty
[ DEFAULT : 7 ]
--pm PM match probability
[ DEFAULT : 80 ]
--pi PI indel probability
[ DEFAULT : 10 ]
--minscore MINSCORE minimum alignment score to report
[ DEFAULT : 50 ]
--maxperiod MAXPERIOD
maximum period size to report
[ DEFAULT : 500 ]
fastqc arguments:
--fastqc FASTQC_PATH path to fastqc
[ DEFAULT : $PATH ]