Potential useful information to review and add to the readthedocs page - from learning unit 2 #365
Labels
Comments
Sign up for free
to join this conversation on GitHub.
Already have an account?
Sign in to comment
This material is from a past lecture and can be reused. It should nevertheless be reviewed.
Types of chromatography (TLC, LC, GC)
Chromatography is used to separate a mixture, e.g. peptides or metabolites, into the individual components. All chromatographic methods have in common, that they have a stationary and a mobile phase. The mobile phase, usually a liquid or a gas, contains the sample and moves through the stationary phase.
Thin layer chromatography (TLC) is a relatively easy, fast and cheap chromatographic technique. It uses a glass or plastic plate which is usually coated with silica gel or aluminium oxide, which acts as the stationary phase. Silica gel is polar which means that polar compounds will interact more strongly with the stationary phase than non-polar compounds.
Liquid chromatography (LC) is the most used family of techniques for separation in proteomics. It comprises all methods that are using a liquid mobile phase and a porous solid as stationary phase. All previously described chromatographic techniques in this chapter are liquid chromatographies.
In the simplest setup, the mobile phase is flowing through the stationary phase under the force of gravity alone. To achieve a result in reasonable time, i.e. an adequate flow rate, high-pressure pumps can be used to move the mobile phase. This implies the name high-pressure liquid chromatography (HPLC).
As HPLC also increases the separation efficiency, HPLC is sometimes interpreted as high-performance liquid chromatography.
Also, the HPLC is the most common separation technique to apply prior to mass spectrometry and most modes can be directly coupled.
Gas chromatography (GC): The column has a small internal diameter of only 0.1 - 0.5 mm and is typically 30 - 60 m long, but depending on the application, may be as long as 120 m. The column is held inside an oven which allows to raise the temperature during the chromatographic separation in a constant and reproducible manner from room temperature to up to 350 °C. Since the separation is based on volatility, this is can be useful to obtain reasonable retention times for compounds which are strongly retained at lower temperatures. One drawback of GC-MS is that only volatile compounds can be measured. To measure non-volatile compounds a tedious derivatization step that increases the volatility is necessary, e.g. polar groups such as OH can be masked.
Separation principles (RP, SAX, SEC)
Reverse phase (RP)
Chromatographic methods using a hydrophobic stationary phase are called reversed-phase chromatography. The term stems from a time where liquid chromatography was commonly performed using a hydrophilic stationary phase, now called normal-phase, in contrast to reversed-phase. Hydrophobic molecules in the mobile phase will tend to bind to the stationary phase, while the hydrophilic molecules pass through and are eluted first.
A hydrophobic stationary phase is generated by modifing the surface silica beads with hydrophobic groups. Most common are carbon chains, as they can be employed in different lengths, corresponding to stronger hydrophobic interactions the longer the chain is. Other carbon configurations are also used, like phenyl groups (carbon rings). C18 is the most frequently used carbon chain for chromatography of peptides.To elute the molecules in order of increasing hydrophobicity, a polar mobile phase with decreasing polarity is employed. A polar mobile phase will drive hydrophobic molecules to adsorb to the stationary phase. As the polarity is decreased, e.g. with a organic solvent, hydrophobic interactions are reduced. As a result, stronger hydrophobic interactions will require a higher concentration of organic solvent in the mobile phase to elute the respective molecules.
Strong Anion Exchange (SAX)
The mode of action for ion-exchange chromatography is based on ionic interactions. Generally, charges alike repel each other and opposite charges attract. These Coulombic forces depend on the charges of two ions and their distance of separation. The analyte is retained by the ionic functional groups on the stationary phase surface with opposite charge. Depending on the type of ionic functional groups on the stationary phase, the type of chromatography gets its name by the ions retained, so either cation (+) exchange or anion (-) exchange chromatography. The more charges an analyte molecule exhibits, the stronger the force acting on it will be. Uncharged molecules will be washed out of the column immideately. The relative amount of cations or anions in the mobile phase will adjust the retention time. There are four established types of ion exchange chromatography: weak and strong cation exchange, and weak and strong anion exhange. When using strong anion exchange the functional groups of the stationary phase are positively charged below pH 10-11.
Size-exclusion chromatography (SEC)
This separation is based on the molecules size, therefore the stationary phase is made of a porous system, acting like a size filter. Provided that all the molecules are loaded simultaneously or near-simultaneously, molecules of the same size should elute together. Different retention times in the elution are ideally influenced solely on the volume of the stationary phase. Small molecules will be able to penetrate every region of the stationary phase pore system, whereas a larger molecule may not penetrate the total volume of the stationary phase, being blocked at some point.This will result in greater path lengths through the stationary phase for smaller molecules. Hence, small molecules will elute late and larger molecules eluting earlier. An important prerequisite is, that the analyte may not interact with the surface of the stationary phase, which would result in retention as a function not only of the size but also of the interaction forces which might be unpredictable.
---------------------- not sure about the graphic here
Peak shapes and properties
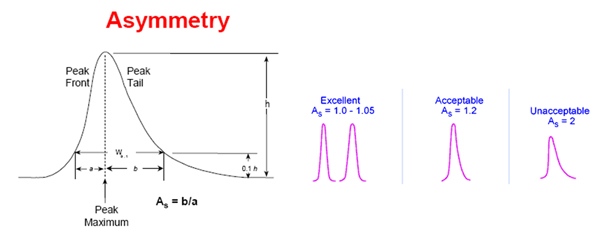
An optimal peak has a gaussian shape but deviations from this may be observed. The asymmetry factor of a peak is given by the distance to the tail of the peak divided by the distance to the front of the peak measured at 10% of peak maximum (apex). An asymmetry factor < 1 is a fronting peak, an asymmetry factor > 1 is a tailing peak (of course small deviations are acceptable).
While for HPLC peak fronting is fairly uncommon many peaks tail to some degree. Tailing occurs mostly due to the interactions with ionized silanol sites on the surface of the stationary phase, although the newer less-acidic type B silica columns reduce this problem, because at a lower pH these sites are fully protonated.
Tandem MS
Determining the mass of an ion is not sufficient to unambiguously identify the peptide or metabolite. Why this is the case can be easily understood for peptides: any permutation of its amino acids result in the same atomic composition and effectively the same mass. Similarly, metabolites with the same sum formula can't be distinguished by solely measuring its mass.
Tandem mass spectrometry is a technique that allows to obtain additional information on the composition of analytes by breaking them apart and measuring its fragments.
In a very common setup, the mass spectrometer determines the m/z of n (e.g. 20) top most abundant moni-isotopic peaks. It then selects and captures ions corresponding to these m/z values for fragmentation. After fragmentation in a collision chamber the fragments are measured in a second mass analysis. Because the selection of ions is performed parallel to the measurement this procedure is called data dependent aquisition (DDA). To prevent redundant fragmentation of the same analyte a so called dynamic exclusion list is usually activated that prevents refragmentation of a m/z ratio for a certain period (e.g. 20 s). This allows the machine to cover a lot more analytes.
The instrument can also be programmed to select only specific ranges of charge states. The restriction to certain charges is very useful as many contaminants are only single charged while peptides typically double or higher charged. It is therefor possible to prevent collection and fragmentation of contaminants like plasticizers.
It is further possible to enter inclusion lists into the machine to specify m/z values for fragmentation. This is often refered to data independent aquisition often used in targeted proteomics / metabolomics.
Fragmentation of gas-phase ions is the process of breaking larger precursor ions (also: parent ions) into constituting parts. If these are charged, they are called fragment ions (also: product ions) that can be measured in a second mass analysis step. The masses of fragment ions obtained in this second analysis step carry valuable information on the composition of the precursor ion. Different fragmentation types exist each giving rise to distinct type of ions. Unfortunately, there is no universal fragmentation method and one has to choose the one that reveals the relevant information on structure or elemental composition of the analytes.
Detail: Modern instruments allow to combine several fragmentation methods to obtain more complete information on the analyte.
Fragmentation can occur during ionisation in the ion source (in-source fragmentation) or later (post-source fragmentation). Nowadays, mainly soft ionisation techniques are used. Accordingly, post-source fragmentation is the predominant fragmentation technique.
Common post-source fragmentation methods are:
Collision-induced dissociation (CID) and Higher-energy collisional dissociation (HCD): In CID and HCD ions collide with neutral atoms or molecules of an inert gas. The part of the mass spectrometer where the collision takes place is called collision chamber. During collision, covalent bonds are broken apart and the ion dissociates typically into two molecules. The charge of the precursor ion distributes on the resulting smaller molecules giving rise to ions but also uncharged molecules. In peptides mainly backbone breaks are induced.Electron transfer dissociation (ETD)
In Electron transfer dissociation (ETD) radical anions are used to transfer electrons to a peptide inducing random breaks at the backbone.
Fragment ions
First, we will take a look at the fragmentation of a single precursor ion and the resulting fragments.
Fragmentation of peptides mainly occur at the backbone. Depending on the fragmentation method the break occurs at different positions and resulting ions are named a,b,c and x,y,z ions.
a and x, b and y and c and z ions are complementary as they correspond to the two ions that can arise from a break at a specific position in a peptide.
a,b anc c ions are the so called prefix ions as they start at the N-terminus of a peptide and reach up to the position where the break took place. x,y and z ions are so called suffix ions as they start at the C-terminus and reach up to the position the peptide was broken apart. The pair of suffix and prefix ion arising from the same fragmentation position are calles sister fragments. The index of a,b,c ions start at the N-terminus counting the residues up to the break. The index of x,y,z ions start at the C-terminus.
Example: The yi
ion is called the sister fragment of the bn−i
ion and vice versa. Both ions can arise from a charged peptide that breaks after the (n-i)-th amino acid.
CID and HCD fragmentation mainly give rise to b and y ions and is the most predominant fragmentation technique.
Detail: ETD fragmentation is different and products are c and z ions.
Immonium ions are a special case of internal fragments. They are composed of a single side chain formed by a combination of a-type and y-type fragmentation.
Internal fragments result from double backbone fragmentation. Usually, these are formed by a combination of b-type and y-type ions, and consist of five residues or less.
Neutral losses occur if an ion looses an uncharged molecule. Most common ones are water loss, ammonia loss and loss of phosphoric acid. Neutral losses can't be measured, of course. But the ion formed by the loss is often seen in tandem mass spectra.
Question: Consider an ion with mass M. What is the mass after neutral loss of water?
On fragmentation, the charge is distributed on one of the resulting fragments. A single charged precursor ion can only give rise to a single charged fragment and a neutral (unobserved) sister fragment. Multiple charged ions can give rise to uncharged, single or multiple charged fragments. The sum of fragment charges is the precursor charge.
We have learned that a single peptide typically breaks into two parts: ion and sister fragment but can also give rise to other types of ions neutral fragments. Fragmenting single peptide ions wouldn't be very interesting as the signal is to weak to be detected. In proteomic and metabolomic mass spectrometry not only one ion is fragmented but many instances of the same ion. The recorded tandem mass spectrum therefor contains an accumulation of signals from many fragmentation events. Because the position of the break differ between the fragmentation events we observe many different ion and sister ion fragments. Ideally, the spectrum of these ions contains masses resulting from breaks at every possible position at the backbone of a peptide. In these case we observe a so called complete mass ladder.
In practice we often have to deal with incomplete mass ladders as some peptides don't tend to fragment at all possible positions.
Similar to incomplete fragmentation the extremer case also exists: some of the precursor ions don't fragment at all and are recorded in the tandem mass spectrum (often with different neutral loses).
Mass accuracy and resolution
So far we have only considered idealized mass spectra containing only pairs of mass-to-charge and intensity. In practice, the mass spectrometer records a raw profile spectrum which contain peaks that resembling a gaussian shape. Turning a profile spectrum into mass-to-charge and intensity pairs is done algorithmically and called peak picking (or centroiding).
The term: full width at half maximum (FWHM) is commonly used in mass spectrometry and defined as the width of a peak at 50% of its maximum height. It is used to describe chromatographic peaks but also of mass peaks.
The mass resolution for a peak made up from singly charged ions of mass m is defined as m/∆m, where ∆m is the FWHM. Note that resolution is dimensionless. Furthermore, in proteomics it has become common to report the resolution for ions at m=400 Th.
Detail: Several definitions of mass resolution exists which also might differ between instrument vendors. Here we only consider the peak width definition.
The text was updated successfully, but these errors were encountered: